Germline variants in cancer therapy
2019-09-09MeikeKaehlerIngolfCascorbi
Meike Kaehler, Ingolf Cascorbi
Institute for Experimental and Clinical Pharmacology, University Hospital Schleswig-Holstein, Campus Kiel, Kiel D-24105, Germany.
Abstract
Keywords: Pharmacogenetics, cytotoxic drugs, anticancer drugs, toxicity, drug resistance, drug metabolism, drug transporter
INTRODUCTION
The tumor cell genome consists of a complex combination of germline and somatic mutations that potentially interfere with anticancer treatment[1].Most genetic mutations in a tumor cell are somatic and are increasingly implicated in targeted therapy with considerable therapeutic benefit.Nevertheless, germline variants may also contribute to interindividual differences in anti-cancer drug response leading to drug resistance, but also to adverse drug effects.Thus, somatic mutations are often associated with treatment efficacy, while hereditary variants are more often considered to address adverse drug effects[2,3].As anticancer treatment often is a double-edged sword balancing best treatment outcome and potential life-threatening adverse drug effects, pharmacogenetic data is relevant to improve individualized therapy.In this context, variants in biotransforming enzymes, drug transporters and their regulators are of major interest as potential biomarkers for improvement of treatment regimen.Here, we discuss the impact of pharmacogenetic testing in treatment with cytostatics and targeted therapies in the context of drug resistance.
CyTOsTaTICs, TaRgeTeD TheRapy aND beyOND
Due to the lack of specific drug targets in various types of cancer and beneficial effects in combination therapy, cytostatics are still of major relevance for anticancer treatment.Metabolism and biotransformation of these compounds underlie various enzymes and transporters, which are well described[4].Hence, hereditary variants in biotransforming enzymes are well known to impair metabolism of drugs, e.g., 5-fluorouracil (5-FU) or 6-mercaptopurine (6-MP).For several cytostatic compounds, pharmacogenetic information has been integrated into treatment recommendations of the Federal Drug Administration or guidelines from the Clinical Pharmacogenetics Implementation Consortium (CPIC), e.g., for 5-FU, irinotecan or 6-MP[5].However, some pharmacogenetic markers still have not been translated into clinical practice.
Within the last 15 years, the number of targeted therapies using small molecules and antibodies drastically expanded with good clinical outcome and increased patient survival.One of the first compounds used was trastuzumab, a HER2/neu-antibody for the use in HER2+ breast cancer[6]and the tyrosine kinase inhibitor imatinib, which targets the BCR/ABL1-kinase in chronic myeloid leukemia[7].In contrast to classical chemotherapeutics, these substances rely on the presence or overexpression of drugable targets on/in the tumor cells.Therefore, expression of the respective target protein in the cancer cell is mandatory and genetic testing of cancer patients is compulsory, e.g., for HER2/neu overexpression for the use of trastuzumab, HER1 for the use of cetuximab or panitumumab or BCR/ABL1/c-kit for the use of imatinib.All of these compounds are dependent on binding to their target protein, which can be impaired by mutation or amplification and by this trigger chemoresistance.While this is a concern in treatment with antibodies and small molecules, drug level of the latter can also be impaired by several other mechanisms.Moreover, intracellular level can be reduced by ATP-binding cassette (ABC) transporters, which facilitate drug efflux or diminished expression or activity of drug importers.Some small molecules are substrates for the CYP450 enzymes, by which the drug plasma level could be impaired.These mechanisms leading to impaired drug level and transport are described below.
heReDITaRy vaRIaNTs IN bIOTRaNsfORmINg eNzymes
Purine analogues, TPMT and NUDT15
The association of thiopurineS-methyltransferase (TPMT) and 6-MP is one of the best documented pharmacogenetic interactions.This purine analogue, as well as azathioprine or thioguanine, are of major relevance in therapy of acute lymphoblastic leukemia (ALL) or non-malignant diseases, e.g., inflammatory bowel disease[8-10].Its anti-inflammatory and anticancer mechanism is based on the generation of active, cytotoxic 6-thioguanosines having major negative impact on leucocyte proliferation.TPMT however prevents the organism from the formation of large amounts of these toxic intermediates through methylation and conversion to inactive methylmercaptopurine.Therefore, patients with TPMT deficiency suffer from a high risk of cytotoxicity due to accumulation of thioguanine nucleotides (6-TGN).Complete absence of TPMT activity may lead to myelosuppression and pancytopenia [Table 1][11].
The frequency of 6-TGN-related hematologic adverse drug events depend on theTPMTgenotype, while intestinal side effects seem to be genotype-independent[12].Nevertheless, pharmacogenetics testing and subsequent adjusting the purine analogue dose is highly recommended[13].
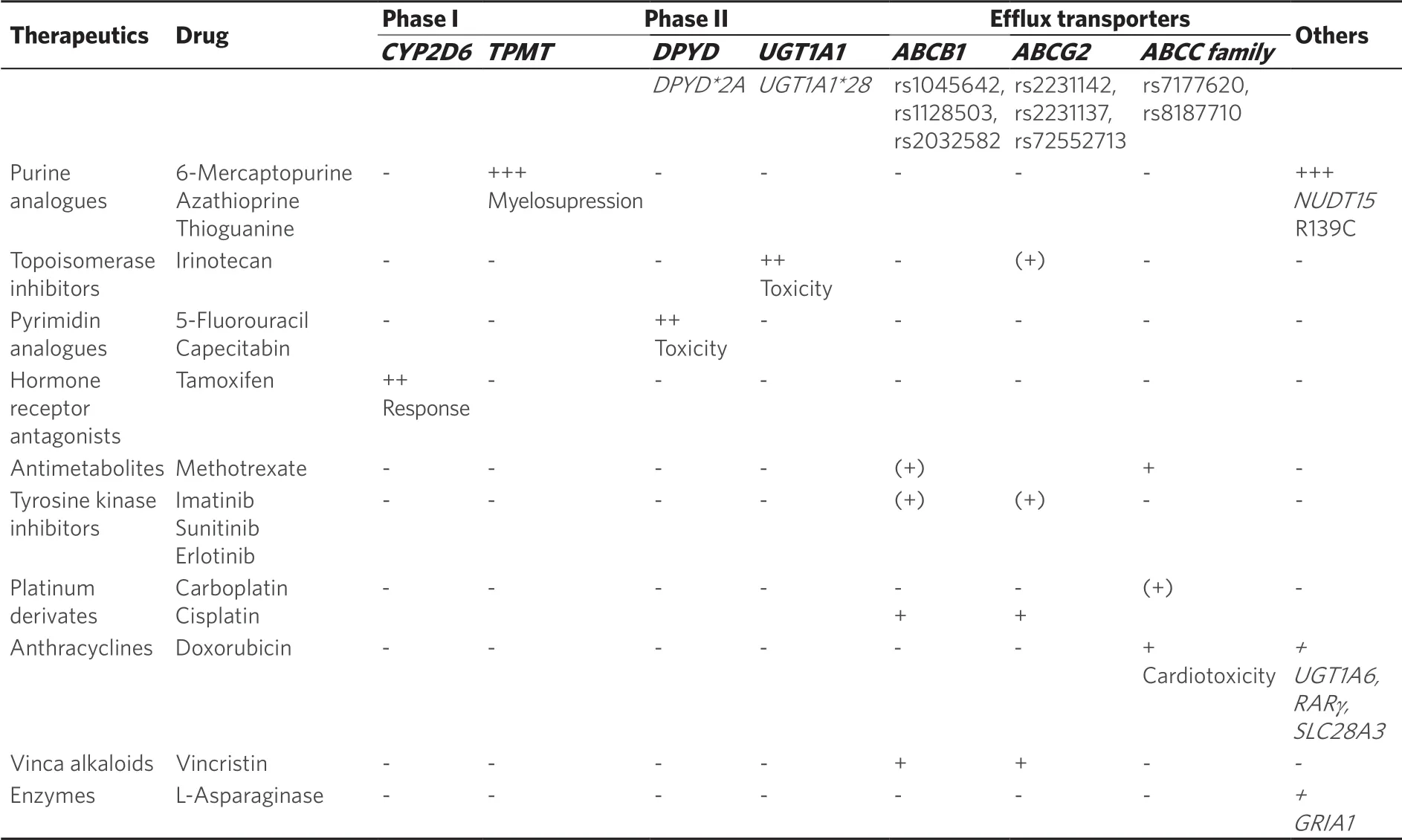
Table 1.Association of hereditary variants in ADME genes to drug-induced adverse effects or therapy response
Only three SNPs, namely ★3A, ★3C and ★2, account for more than 90% of TPMT inactivating variants in the Caucasian population[13,14].According to theTPMTgenotype, the phenotype is distinguished into normal, intermediate and poor metabolizers.Some diplotypes are still considered as “undetermined” such asTPMT★1/★8or★6/★8.The most recent CPIC guideline suggests starting dose reductions of 50%-80% for immediate, while the starting dose in poor metabolizers should be drastically reduced to 10% and administered thrice weekly instead of daily[23].The dose should be carefully titrated depending on the grade of myelosuppression[15].
Interestingly, TPMT seems to be of particular relevance in Caucasian, while in Asian populationTPMTvariants are less frequent, but the degree of leukopenia is more pronounced[16,17].In Asian populations, another polymorphic gene was discovered to play an important role in thiopurine toxicity.Yang and coworkers identified a significant association of the NUDT15 (nucleoside diphosphate-linked moiety X type motif 15) variant p.R139C to thiopurine-related leukopenia, which is involved in nucleoside diphosphate metabolism, especially built in stress response[18].In this study among children with ALL, those being homozygote for the variant allele p.R139C tolerated only 8% of the standard mercaptopurine dose.In contrast, the tolerated dose intensity in heterozygous and wildtype carriers was 63% and 83.5%, respectively.As NUDT15 catalyzes thiopurine inactivation to thioguanine monophosphate, the almost complete loss of NUDT15 activity led to accumulation of thiopurine-metabolites causing toxicity [Table 1][19-21].Meanwhile further variant alleles have been identified with varying prevalence among different ethnicities and varying and sometimes uncertain functional effects.A recommendation of aNUDT15nomenclature was published very recently by the Pharmacogene Variation (PharmVar) Consortium[22].
Due to the high clinical significance ofNUDT15gene variation especially for Asian populations, the CPIC guideline for thiopurine dosing was amended[23].To avoid thiopurine toxicity both,TPMTas well asNUDT15genotypes should be considered, although theNUDT15genotype-related dose recommendations are not yet fully clear.
lrinotecan and UGT1A1
N- orO-glucuronidation by specific UDP-glucoronosyltransferases (UGT) is a crucial step in the elimination process of a number of drugs and endogenous compounds.Usually such conjugation elevates the compound's hydrophilicity thereby facilitating excretion into urine.This is exemplified by UGT1A1 that catalyzes the transformation of bilirubin to bilirubin-glucuronide in the liver, before this conjugate can be secreted via the renal ABC transporter ABCC2 into urine.
In this context,UGT1A1★28 (rs8175347), a TA tandem repeat in theUGT1A1promoter causing Gilbert syndrome/Morbus Meulengracht is of relevance causing defective heme metabolism.Wild type alleles harbor six TA repeats, whereas the seven TA repeat variant leads to diminished protein expression.Although presence ofUGT1A1★28 is abundant in Caucasians (minor allele frequency of carriers 40%), this genetic variant itself does not contribute to development of defective heme metabolism, as frequency of Gilbert syndrome is only 3%-9% in Europeans[24-27].
The topoisomerase I-inhibitor irinotecan, widely used for the treatment of colorectal cancer in combination with 5-FU, is one of the drugs being metabolized by UGT1A1.Carriers ofUGT1A1★28 variants bear a higher risk for drug toxicities as UGT1A1 is required for detoxification of the drug.Low UGT1A1 activity may result in diarrhea and neutropenia in a dose-dependent manner [Table 1][28].So far,UGT1A1★28 genotyping is recommended as part of the irinotecan drug label, although no distinct doses are given.Dependent on the dosage, patients being heterozygous carriers should be closely monitored for any toxicities, while for homozygous carriers the starting dose should be reduced[15,29].Recently, a study in Japanese colorectal cancer patients revealed that reduction of the initial irinotecan dose from 150 mg/m² to 120 mg/m² results in a safe and efficient therapy inUGT1A1homozygote variant allele carriers[30].It should be noted that among Asian populations, c.211G>A leading to p.Gly71Arg and allocated toUGT1A1★6should be considered as well due the low activity phenotype.A retrospective study from Switzerland however could not confirm thatUGT1A1★28is the only risk factor for neutropenia and diarrhea.In a multifactorial analysis baseline neutrophil count, sex, age and performance status were additional items contributing to the complexity of adverse events[31].Studies investigating the question, whether elevated irinotecan doses are tolerable among wild-type carriers are ongoing.Here, pharmacogenetic testing could contribute not only to the avoidance of toxicity, but also to a putative better clinical outcome of the disease.
Pyrimidine analogues and dihydropyrimide dehydrogenase
The pyrimidine analogue 5-FU is commonly used for treatment of metastatic colon carcinoma or breast cancer.Main metabolism of the drug is performed in the liver by dihydropyrimide dehydrogenases (DPD), which also metabolize other fluoropyrimidines, e.g., capecitabin.Defects in DPD lead to accumulation of 5-FU and systemic toxicity, ranging from myelosuppression, neurotoxicity or diarrhea [Table 1].
For DPD, several rare variants have been described (also reviewed in[32]):DPYD★2A(rs3918290) leads to the formation of an alternate splicing side in intron 14, resulting in expression of a 165 bp-deletedDPYDmRNA and protein lacking the amino acid residues 581-635[33].The AA variants leads to complete DPD deficiency and severe 5-FU toxicities[34].Especially in African-Americans,DPYD★9A(rs1801265) has been described in 13%-39% of the population[35].The effect of this variant is still controversial, as some studies revealed association ofDPYD★9Agenotype and 5-FU side effects in gastrointestinal malignancies[36], but others did not observe impairment of enzyme activity of the 9A haplotype[37].Overall, this points to a dependency of this haplotype to other non-genetic factors.
For further rare genetic variants with low frequencies, especiallyDPYD★3(rs72549303) andDPYD★4(rs1801158), analyses of effects and dose-adjustments is still ongoing.In anin vitro-expression system, reduced enzyme activity was observed for D949V, while M166V, E828K, K861R and P1023T variants leading to increased enzyme activity compared to the wild type[35,38].
DPD activity scores are based on the functionally characterized variants c.190511G>A, c.1679T>G, c.2846A>T, and.c.1129-5923C>G.Based on these estimated activity scores, the recent CPIC guidelines strongly suggest for patients with complete DPD deficiency to avoid the use of fluoropyrimidines[39].This is also due for patients having a very low activity score of 0.5.In cases were no alternative for 5-FU are considered, strong dose reduction and therapeutic drug monitoring is recommended.Since also intermediate metabolizers have an increased risk of adverse events, dose reductions of 25%-50% should be considered.
Moreover, combinational analysis ofDPYDandUGT1A1genotypes in Italian colorectal cancer patients receiving fluoropyrimidines/irinotecan revealed that the incremental cost betweenDPYDvariant andUGT1A1★28/★28carriers and non-carriers was €2,975, indicating the relevance of these pharmacogenetic traits also from an economic perspective[40].In addition, a recent study stressed the relevance ofDPYDtesting accompanied by genotype-dependent dose reduction for the use of fluoropyrimidines[41].Concluding, patients not only of non-European descent could benefit from genetic testing forDPYD/UGT1A1variants.
Tamoxifen and CYP2D6
The estrogen receptor antagonist tamoxifen has been successfully used in treatment of estrogen-receptor positive breast cancer since more than 40 years.Tamoxifen is metabolized mainly by CYP2D6 resulting in various metabolites of which endoxifen has the strongest affinity to the estrogen receptor.As a result,CYP2D6genotype tightly correlates to endoxifen plasma levels in patients.In several studies it was shown thatCYP2D6genotype is a determinant marker of tamoxifen efficacy [Table 1] contributing to 34%-52% of absolute endoxifen plasma level[42].Several variants have been described forCYP2D6being associated with normal (CYP2D6★1,CYP2D6★2), decreased (e.g.,CYP2D6★10) or complete loss of function (e.g.,CYP2D6★3,CYP2D6★4).Furthermore,CYP2D6copy number variants may lead to excessive metabolism and ultrarapid CYP2D6 phenotype (reviewed in[43]).Indeed, several studies suggested an association ofCYP2D6genotype and overall survival, relapse- or recurrence-free survival[43-46].Accordingly, the CPIC guidelines forCYP2D6and tamoxifen therapy recommend that CYP2D6 poor metabolizers should be treated alternatively with aromatase inhibitors and in CYP2D6 intermediates dosage adjustments should be performed to prevent therapy failure of tamoxifen.In addition, CYP2D6 inhibitors should be avoided regardless of theCYP2D6genotype[43].Regarding the use of treatment alternatives, esp.aromatase inhibitors, their use in premenopausal patient still is contraindicated due to occurrence of treatment-associated amenorrhea, unless suppression of ovarian function is performed simultaneously[47].This has been shown in recent studies, clearly showing superior overall and disease-free survival rates in combination of aromatase inhibitors or tamoxifen plus ovarian suppression[48].In post-menopausal patients, the optimal treatment strategy is still controversial, as treatment-duration and -switching between tamoxifen and aromatase inhibitors is still under investigation[49,50].This adds additional complexity to the relevance ofCYP2D6genotyping for the use of tamoxifen.So far, due to a number of inconsistencies, pharmacogenetic analysis ofCYP2D6status has not yet been considered in the drug label.As reviewed recently, some inconsistencies in the role ofCYP2D6can be explained by the proportion of DNA isolated from peripheral blood cells or tumor tissue.So far, any association of treatment outcome toCYP2D6genotype failed when using tumor tissue as DNA source[51].However, in 960 women from the Quilt Study cohort, neither theCYP2D6genotype nor the concomitant use of CYP2D6 inhibitors had any influence on the clinical outcome[52].In addition, in a prospective study including 667 pre- and postmenopausal patients, no association was found between endoxifen concentrations and relapse-free survival time, and there was also lack of association betweenCYP2D6genotype and relapsefree survival time.As any selection bias or non-consideration of confounding factors was regarded as small, the authors concluded, that the results of this study do not supportCYP2D6genotyping to guide tamoxifen treatment in the adjuvant setting[53].
Promising variants as new predictive factors
Within the last years, several studies revealed novel promising candidates hereditary variants apart from ADME genes that might help to predict ADE under chemotherapy regimen.For these, we would like to stress some recent findings in anthracycline-derived cardiotoxicity andL-asparaginase-induced hypersensitivity reactions.
Anthracylines, e.g., doxorubicin or daunorubicin, are of major relevance for the treatment of multiple solid tumors and leukemia.Severe side effects, in particular cardiotoxicity occurring in about 57% of all patients is a frequent dose limiting cause.In the last years, several studies have shown hereditary variants in UDPglucuronosyltransferase 1A6 (UGT1A6; taq marker ofUGT1A6★4, rs17863783), the nucleoside transporterSLC28A3(L461L, rs7853758; intronic variant, rs885004) or retinoic acid receptor gamma (RARγ; rs2229774) as promising predictive biomarkers (comprehensively reviewed in[54]).The silent variant V209V (rs17863783) inUGT1A6was associated with increased risk of anthracycline-induced cardiotoxicity in several patient cohorts, potentially resulting in decrease of enzyme function and accumulation of toxic metabolites[55,56].A similar phenomena has been shown for the sodium-coupled nucleoside transporterSLC28A3, as rs7853758 and rs885004 might interfere with enzyme activity promoting risk of cardiotoxicity[57].In addition, the amino acid exchange S427L (rs2229774) in retinoic acid receptor gamma (RARγ) was found in a GWAS study to be associated with cardiotoxic risk increase in children[58].Earlier studies reported an association ofABCC1efflux transporter variants with doxorubicin-associated cardiomyopathy[59].However, so far the predictive value of these variants is not determined or low, preventing its implementation into clinical practice so far.
Regarding the use ofL-asparaginase in ALL, hypersensitivity reaction have been observed.Interestingly, pharmacogenetics studies revealed an association to multiple polymorphisms (rs4958351, rs10070447, rs6890057, rs4958676, rs6889909, rs11167640, rs10072570, rs13354399, rs17356099, rs2055083, rs707176) in the AMPA 1 glutamate receptor (GRIA1)[60,61].These data clearly point to inherited factors contributing to treatment hypersensitivity, however, functional validation is necessary to understand the relevance of these variants.
Finally, the treatment of solid tumors with taxanes, such as paclitaxel, docetaxel, and cabazitaxel is commonly associated with treatment-limiting peripheral sensory neuropathy.Currently there are no biomarkers available, allowing a precise prediction of such chemotherapy-induced peripheral neuropathy (CIPN)[62].The attempt to reveal predictive genetic markers through GWAS studies disclosed an intronic SNP (rs875858, MAF ¼ 0.056), in theVAC14gene.VAC14 encodes a scaffold protein that is a component of the PIKfyve protein kinase complex.Interestingly this complex is involved in the synthesis of phosphatidylinositol 3,5-bisphosphate.Knock-out experiments in mice caused severe neurodegeneration[63].The authors of the GWAS study further made mechanistic approaches to validate the functional consequences of theVAC14variant.iRNA knockdown ofVAC14in stem cell-derived peripheral neuronal cells increased docetaxel sensitivity andVAC14heterozygous mice had greater nociceptive sensitivity than wild-type controls.Further GWAS meta-analyses of taxane-related CIPN were however not able to identify significant associations of any genetic variant to ≥ grade common terminology criteria for adverse events (CTCAE) neuropathy in European and African Americans after correction for multiple testing[64].Concluding, there are some interesting genetic risk candidates, however their suitability for predicting treatment-limiting peripheral sensory neuropathy has not been confirmed so far.
mUlTIDRUg ResIsTaNCe IN CaNCeR TReaTmeNT aND ITs phaRmaCOgeNeTIC CONTRIbUTIONs
The term “multidrug resistance” is tightly linked to expression of ABC-transporters in cancer cells[65].By efflux of substrates across the membrane, the intracellular level of the drug is lowered and drug resistance becomes more likely.Furthermore, bioavailability of drugs is impaired influencing adsorption, distribution and elimination.The most prominent transporters regarding chemoresistance are P-glycoprotein (P-gp, ABCB1), breast cancer resistance protein (BCRP, ABCG2) and members of the ABCC-family, especially ABCC1/MRP1 (multidrug resistance protein 1), ABCC2/MRP2 (multidrug resistance protein 2) and ABCC3.Drug resistance via drug transporters is not only influenced by deregulation of gene expression, but also by genetic variants[66].While a vast number of studies showed impact of genetic variants on drug transporter expression or activities, recent data dealt with variation of 3'-UTR length in drug resistant cancer cells.
P-gp in drug resistance
A huge number of classical and targeted chemotherapeutics are substrates for P-gp and many studies demonstrated an overexpression of P-gp upon exposure to such drugs [Table 1].Consequently, a potential impact of genetic variants onABCB1expression and subsequently in response to treatment could be found with more than 4,453 SNPs identified in this gene.Most clinical studies focused on 3435C>T, 2677G>T/A and 1236C>T that were suggested to impair P-gp expression or activity.
The cytosine deamination at position 3435 (rs1045642) in exon 26 does not result in an amino acid substitution.Nevertheless, a number of studies suggested a lower P-gp expression and reduced P-gp function in 3435T carriers[67,68].In acute lymphocytic leukemia, carriers of the wildtype revealed higher toxicity after high-dose methotrexate treatment, while the risk of relapse was reduced in carriers of at least one variant allele[69].Nonetheless, a direct association to cancer therapy failure could not be seen.The impact of the silent SNP 1236C>T (rs1128503) in exon 12 instead is not entirely understood, as contradictory studies showed opposite results regarding response to imatinib in chronic myeloid leukemia[70,71].In the same studies, amino acid substitution of alanine to serine/threonine due to 2677G>T/A (A893T, rs2032582) could be associated to imatinib response, with contradictory effects as carriers of the CC variant revealed altered susceptibility towards imatinib therapy compared to wildtype carriers.Regarding missense mutations inABCB1, an experimental approach usingSaccharomyces-based assay revealed several non-synonymous SNPs in the context of chemoresistance, e.g., 266T>C (M89T), 1985T>G (L662R), 2005C>T (R669C) 3322T>C (W1108R) and 3421T>A (S1141T)[72,73].Nevertheless, clinical relevance of these SNPs has to be confirmed.Overall, the issue onABCB1variants and their association to drug response remains controversial.Due to its high phenotypic variability however, it is more than questionable to useABCB1variants to as predictive biomarker in cancer therapy.In addition to hereditary variants, the regulation of ABC transporters by nuclear receptors, cytokines and also microRNAs is of major importance.In this regard, differential expression of 3'-UTR lengths of theABCB1mRNA might additionally contribute to P-gp variability (see epigenetics section).
ABCG2/BCRP and chemoresistance
Besides P-gp, BCRP was identified as a contributing factor to multidrug resistance [Table 1].Being highly expressed in hematopoetic precursor and stem cells[74], it is regarded as a stem cell factor[75].Moreover, its high expression in tumor cells pointed to a potential role in chemoresistance and therapy failure[76,77].Main research has been performed on non-synonymous 34G>A and 421C>A.The SNP 34G>A (rs2231137) results in exchange of valine to methionine (V12M) at the N-terminus potentially leading to reduced protein expression.This SNP could be associated to outcome of tyrosine kinase inhibitor therapy, especially using imatinib[78].Some clinical evidence point to a better response to imatinib in CML patients carrying the homozygous variant[78].A similar tendency was observed in metastatic renal cell cancer treated with sunitinib[79].On the contrary, an association of therapy using erlotinib in B cell lymphoma patients could not be observed[80].The 421C>A substitution (rs2231142) leads to an amino acid exchange from glutamine to lysine (Q141K) that affects the ATP binding domain.In a number of studies, bioavailability of drugs, e.g., the topoisomerase inhibitor topotecan[81]or the tyrosine kinase inhibitor imatinib[82], was impaired in carriers of the 421C>A genotype.Regarding tyrosine kinase inhibitors, many compounds are substrates of BCRP, however transport is dose-dependent[83].Interestingly, the 421A haplotype could be associated to modulate posttranscriptional regulation via the 3'-UTR[84].Nevertheless, no significant association of 421C>A was found or results were contradictory.Besides these two SNPs, 376C>T leads to formation of a premature stop codon (Q126stop, rs72552713) resulting in diminished protein expression and could be associated to drug hypersensitivity[85].Conversely, the promoter SNP -15994C>T results in increased BCRP expression and imatinib clearance[86].
The ABCC subfamily and their genetic variants in chemoresistance
For the ABCC subfamily, twelve members have been described yet.With regards to chemoresistance ABCC1, ABCC2 and ABCC3 have been intensively investigated.While anticancer drugs of the new generation are mainly transported by ABCB1 and ABCG2, several cytostatics, e.g., vincristine or cisplatin, are substrates for these transporters [Table 1][87].As distinct clinical studies on association of ABCC1 and ABCC3 to drug resistance are lacking, most data is present on ABCC2.Most prominent SNPs are -24C>T in theABCC2promoter region (rs7177620) being associated to reduced expression and activity of the protein[68].This could thus be linked to higher methotrexate plasma levels[88].Together with 1249G and 3972T, -24T haplotype was associated with imatinib resistance[89].Regarding therapy using platinum or platinum-derivates, e.g., carboplatin, it was observed that 4544G>A (rs8187710) correlated with adverse survival rates of patients suffering from non-small cell lung cancer[90].Furthermore, this SNP was associated with doxorubicininduced cardiotoxicity requiring closer monitoring during treatment[59].
Chemoresistance, epigenetics and miRNA regulation
It is well known that protein expression can be modulated on the transcriptional level by epigenetics, as well as post-transcriptionally by microRNAs or RNA-binding proteins.Analyses of epigenetics, e.g., DNA methylation or histone modification, remain inconclusive for ADME genes so far.In several studies, data on promoter methylation of the efflux transportersABCB1orABCG2showed contrary differential methylation in various types of cancer[91-96].In addition, hereditary variants in promoter regions might directly impair binding of transcription factors leading to differential gene expression.This has also been shown for several cytochrome P450 enzymes pointing to gene expression regulation by methylation, e.g., CYP1A2, CYP2C19 and CYP2D6[97,98].
Main research has been performed for transcriptional control by microRNAs resulting in blockade of translation or degradation of their respective target mRNAs[99].This interaction can be impaired by hereditary variants of the mRNA 3'-UTRs in target genes.For ABC-transporters, several microRNA interactions have been described so far, e.g., miR-508-3p/ABCB1, miR-212/ABCG2or miR-379/ABCC2[91,100,101].For the latter, it was observed thatABCC2-24C>T, 1249G>A and 3972C>T variant haplotypes led to pronounced microRNA-dependent suppression compared to wild-types[102].
Interestingly, the interaction of microRNA/mRNA can also be impaired by selective expression of alternative mRNA 3'-UTRs by the cancer cell.As these shorter 3'-UTRs lack the microRNA-binding regions, regulation of respective microRNA can be abolished.This phenomenon was observed not only forABCG2in mitoxantrone-resistant colon cancer cells, but also forABCB1in chronic myeloid leukemia cells[103,104].Potentially, this is also relevant for other ABC transporters or biotransforming enzymes (reviewed in[105,106]).Further studies are required to investigate this mechanism and its regulation.
Within the last decade it was observed that microRNA expression pattern are distinct for each tumor and can be altered during in treatment with anticancer drugs (reviewed in[107]).Thus, haplotype-dependent microRNA interaction and regulation of mRNA expression might contribute to differential susceptibility of tumors and patients.
CONClUsIONs aND OUTlOOK
Pharmacogenetic testing of somatic and germline mutations is an emerging field in oncology.Presence of hereditary variants are relevant for avoidance of adverse drug events in some classic chemotherapy regimen, such as thiopurines, 5-FU or irinotecan.Pharmacogenetic testing for hereditary variants in metabolic enzymes likeTPMTandNUDT15,UGT1A1orDPYDmight be helpful to prevent patients from severe adverse drug effects by subsequent adjustment of the drug dose or therapy alternatives.Although genetic testing for these traits is not mandatory, implementation into the respective drug label has been performed and genetic testing is recommended.Overall, testing for hereditary variants in these genes might be helpful to predict adverse drug effects and improve patients' compliance.
In contrast to variants in biotransforming enzymes, association of SNPs in drug transporters to the clinical outcome is controversially discussed.Evidence on the predictive value of many SNPs on the respective gene expression, function and association to drug efficacy is widely lacking.Therefore, the impact on hereditary variants in ABC transporters on overall therapy response remains questionable.
Overall, pharmacogenetic testing of single traits might not be sufficient to explain neither interindividual differences, nor intratumoral differences in many cases, as these underlie clonal evolution.Within the last decades, there is much more understanding of tumor heterogeneity and complexity and research has been widely evolved.Technologies such as whole genome analyses, RNA-seq or epigenetic analyses have been shown to be crucial to gain deeper insights into the complexity of tumor traits and to design novel therapy strategies and co-treatments.Especially for targeted therapy regimen, testing for presence of drugable targets in or on tumor cells is mandatory (e.g., BCR/ABL1 or HER2).Moreover accompanied diagnostics of tumor mutations is required for an increasing number of anticancer drugs including biologicals.Furthermore, the invention of checkpoint blockade inhibitors like ipilimumab, nivolumab and pembrolizumab leading to reactivation of T cell anti-tumor response revolutionized treatment in a variety of malignancies[108-110].For these compounds, tumor dependency on the respective target is necessary for therapeutic benefit.
Nevertheless, co-integration of genetic testing into the clinical routine for somatic and germline variants in cancer simultaneously could be helpful to optimize therapy regimen and improve patients' compliance and survival.
DeClaRaTIONs
Authors' contributions
Made substantial contributions to conception and design of this review, performed data analysis and interpretation and wrote the manuscript: Kaehler M, Cascorbi I
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2019.
杂志排行
Cancer Drug Resistance的其它文章
- Alkylating anticancer agents and their relations to microRNAs
- Pharmacogenetics implementation in the clinics: information and guidelines for germline variants
- lnfluence of lysosomal sequestration on multidrug resistance in cancer cells
- Opportunities and challenges of implementing Pharmacogenomics in cancer drug development
- Pharmacogenetics of anticancer monoclonal antibodies
- Resistance to anti-tubulin agents: From vinca alkaloids to epothilones