Methionine adenosyltransferases in liver cancer
2019-09-02BenMurrayLuciaBarbierTorresWeiFanJosMatoShellyLu
Ben Murray, Lucia Barbier-Torres, Wei Fan, José M Mato, Shelly C Lu
Abstract Methionine adenosyltransferases (MATs) are essential enzymes for life as they produce S-adenosylmethionine (SAMe), the biological methyl donor required for a plethora of reactions within the cell. Mammalian systems express two genes,MAT1A and MAT2A, which encode for MATα1 and MATα2, the catalytic subunits of the MAT isoenzymes, respectively. A third gene MAT2B, encodes a regulatory subunit known as MATβ which controls the activity of MATα2.MAT1A, which is mainly expressed in hepatocytes, maintains the differentiated state of these cells, whilst MAT2A and MAT2B are expressed in extrahepatic tissues as well as non-parenchymal cells of the liver (e.g., hepatic stellate and Kupffer cells). The biosynthesis of SAMe is impaired in patients with chronic liver disease and liver cancer due to decreased expression and inactivation of MATα1. A switch from MAT1A to MAT2A/MAT2B occurs in multiple liver diseases and during liver growth and dedifferentiation, but this change in the expression pattern of MATs results in reduced hepatic SAMe level. Decades of study have utilized the Mat1a-knockout (KO) mouse that spontaneously develops non-alcoholic steatohepatitis (NASH) and hepatocellular carcinoma (HCC) to elucidate a variety of mechanisms by which MAT proteins dysregulation contributes to liver carcinogenesis. An increasing volume of work indicates that MATs have SAMe-independent functions, distinct interactomes and multiple subcellular localizations. Here we aim to provide an overview of MAT biology including genes, isoenzymes and their regulation to provide the context for understanding consequences of their dysregulation. We will highlight recent breakthroughs in the field and underscore the importance of MAT's in liver tumorigenesis as well as their potential as targets for cancer therapy.
Key words: Methionine adenosyltransferases; S-adenosylmethionine; Liver cancer;Hepatocellular carcinoma; Cholangiocarcinoma; Biomarkers; Therapeutic targets and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See:http://creativecommons.org/licen ses/by-nc/4.0/
INTRODUCTION
This review examines the roles of methionine adenosyltransferases (MATs) in liver cancers with a focus on how dysregulation of these proteins contributes to their pathogenesis. Hepatocellular carcinoma (HCC) is the most common primary liver cancer and currently the second leading cause of cancer-related death worldwide[1]. In the majority of the cases, HCC develops in patients with underlying chronic liver disease and cirrhosis, which are mainly derived from viral hepatitis infection, alcohol abuse and non-alcoholic fatty liver disease (NAFLD)[2]. The threat of HCC is expected to continue rising due to increasing cases of NAFLD given the obesity epidemic occurring worldwide[3]. Even more alarming are reports of HCC from NAFLD that occurred in the absence of cirrhosis[4]. Cholangiocarcinoma (CCA) is the second most common primary liver cancer, which typically occur in the setting of chronic biliary inflammation. MATs exert very similar roles in both types of liver cancers.
OVERVIEW OF MAT ENZYMES
MAT genes and isoenzymes
MATs (EC 2.5.1.6) belong to a family of enzymes th at are essential to life as they catalyze the biosynthesis of SAMe, the main methyl donor of the cell. Apart from methylation, SAMe is also important as a precursor in glutathione (GSH) and polyamine synthesis[5]. As SAMe is used for the methylation of biomolecules including DNA, RNA, proteins, biological amines and phospholipids, any change in its biosynthesis and catabolism within the cell has a profound effect on cellular processes such as growth, differentiation and response to injury. Over the past decade SAMe-independent roles of MAT enzymes have emerged, most notably, their ability to function as bona fide transcription factors as well as their ability to form parts of scaffold complexes which have been implicated in cancer development[6-9].
In mammals, three distinct genes, MAT1A, MAT2A, and MAT2B give rise to the protein products MATα1, MATα2, and MATβ, respectively (Table 1, Figure 1)[10].MAT1A encodes for a 395-amino acid (396 in mouse, and 397 in rat) catalytic subunit in humans that is mainly expressed in hepatocytes (parenchymal cells of the liver) as well as bile duct epithelial cells and pancreatic acinar cells[8,11]. MATα1 can oligomerize to form a homotetramer (MATIII) or homodimer (MATI). The MATα2 subunit, the extrahepatic catalytic protein (395 amino acids), that has an 84% sequence identity to MATα1, can also form dimers and tetramers but with a bias towards the dimer conformation[10,12,13]. MATα2 is expressed in non-parenchymal cells of the liver as well as in all extrahepatic tissues. The regulatory subunit MATβ has four known isoforms, MATβV1, MATβV2, MATβV2a, and MATβV2b with the former two being the major splice variants[14]. MATβV2a and MATβV2b are expressed at very low levels compared to MATβV2 and have not been studied in detail[14]. MATβV1 is expressed in fetal liver, prostate, lung, brain, thyroid and the adrenal gland, whilst MATβV2 is expressed in skeletal muscle and heart. Both isoforms are expressed in the kidney and thymus[14]. MATβV1 and MATβV2 are 331 and 323 amino acids long, respectively,differ only by 20 amino acids at their N-terminus and share little sequence identity(7%) to the catalytic MAT proteins. MATβ has a binding pocket for the cofactor NADP+, which can interact with both major isoforms[12,15].
MAT expression patterns and subcellular localization

Table 1 Mammalian MAT genes and isoenzymes
MAT enzymes were first thought to function as SAMe producing factories in the cytosol, and that SAMe would be delivered to the specific compartments such as the nucleus or mitochondria for methylation reactions[16]. However, a decade ago MATα1 was reported to be present within the nucleus[17]. This was followed by publications describing MATα2, MATβV1 and MATβV2 to also be within this organelle[6,18]. These publications showed that a nuclear location of MAT proteins was associated with enhanced histone H3K27 methylation, an epigenetic modification that leads to gene silencing[17]. Most recently MATα1 was reported to be present in the mitochondrial matrix of hepatocytes, enhancing mitochondrial function and negatively regulating cytochrome P450 2E1 (CYP2E1) through methylation[19]. These accumulating publications reinforce the concept that MATs are recruited to subcellular compartments to provide a local source of SAMe.
Structural overview of MATs and MAT complexes
To date there are numerous crystal structures of MAT proteins from different species in different active site conformations, complexed with substrates, products, or their analogues[12,20-23]. The catalytic proteins have a three-domain organization that is conserved amongst other MAT family subunits[21,22,24]. Monomeric MAT enzymes are incapable of producing SAMe as they do not contain a complete active site. Upon dimerization, both monomers contribute residues to form two active sites. The large hydrophobic surface of monomeric MAT constitutes the site of the monomermonomer interface. A common feature of MAT enzymes is that they contain a “gating loop” (in human MATα1 and α2 residues 113-131) that flanks the active site, which has been hypothesized to move dynamically to allow access to the active site[21]. When the active site is occupied, the loop closes to form a gate over the active site, but when the active site is empty, the loop becomes disordered or open. MATα1 dimers can also form tetramers through the central domain of the subunits[24]. Recent work has shown that mutation to residues of the gating loop can reduce enzyme activity and SAMe formation[25].
The regulatory βsubunit is structurally very different from the αsubunits, unable to produce SAMe by itself. MATβ proteins contain signature motifs of the SDR (shortchain dehydrogenase/reductase) superfamily including a Rossman fold that can bind FAD+or NADP+although MATβ favors the latter[22]. MATα2 and MATβ interact to give rise to the MATα2β complexes (also referred to as MATII)[12,26](Figure 1, Table 1).To date only the structure of a MATα2βV2complex has been solved and it consists of a MATα2 tetramer flanked by two MATβV2 subunits (MATα2(4)βV2(2)). This showed that MATα2 can exist and function as a tetramer in the presence of MATβ[12]. The oligomeric state of this crystal structure, confirmed with small angle x-ray scattering[12], is different from the suggested tetrameric form [MAT(α2)2(β)2][27]or the proposed computational model in which MATαβ was assumed to be a trimer[MAT(α2)2(β)1][15]. Mutational analysis showed by gel filtration that the minimum motif required for the formation of the MAT(α2)4(βV2)2complex comprises three residues at the end of the C-terminus of MATβV2 (V321F322H323)[12]. Several publications,using recombinant purified proteins, have shown that MATα1 can also interact with MATβV1, although this interaction is several orders of magnitude weaker than that of MATα2 and MATβ[12,25]. The MATα1βV1 complex is not likely to occur within the cell as MATα1 and MATβ are generally not expressed at the same time.
MAT enzymatic mechanism
The wealth of structures available for MAT enzymes, as well as the range of biochemical evidence, has provided terrific detail and insight into the enzymatic mechanism. SAMe is produced by the addition of the amino acid methionine to the energy molecule ATP (Figure 2). Upon entry of the substrates ATP and methionine into the active site, the flexible gating loop becomes well-ordered closing the active site. The synthesis of SAMe follows an SN2catalytic mechanism[28,29], whereby the sulphur atom of methionine attacks the C5' atom of ATP displacing the tripolyphosphate (PPPi) moiety to form SAMe. The PPPi is then hydrolyzed giving rise to pyrophosphate (PPi) and orthophosphate (Pi), providing the energy to facilitate product release by dislodging the gating loop[30]. For a detailed mechanism, see Komoto et al[21]2004 and Murray et al[23]2016.
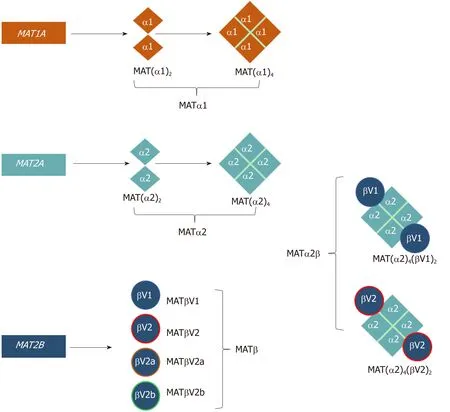
Figure 1 Schematic representation of the oligomeric states of mammalian MAT enzymes.MAT1A and MAT2A genes encode the catalytic subunits MATα1 and MATα2, respectively. Both MATα1 and MATα2 can be organized as dimers and tetramers. MAT2B encodes the regulatory subunit for which there are four isoforms, MATβV1, MATβV2,MATβV2a, and MATβV2b with the former two being the major splice variants. MATα2 and MATβ interact to give rise to the MATα2β complexes. The MAT(α2)4(βV1)2 and MAT(α2)4(βV2)2 complexes consist of a MATα2 tetramer flanked by two MATβV1 or MATβV2 subunits.
MAT activity
Despite high sequence identity MAT enzymes exhibit different kinetic and regulatory properties for methionine, ATP, and SAMe. The Kmfor the methionine is lowest in MATα2 followed by MAT(α1)2and is the highest for MAT(α1)4[5]. The Kmfor ATP is also highest for MAT(α1)4(1-2 mmol), intermediate for MAT(α1)2(0.2-0.5 mmol), and lowest for MATα2 (70 μM)[31,32]. SAMe, the product produced by MAT, can act as a feedback inhibitor to some of these enzymes[33]. MATα2 is the most sensitive to SAMe with a 50% inhibitory concentration (IC50) of 60 μM which equates to the normal physiological liver levels. MAT(α1)4is minimally inhibited by SAMe (IC50= 400 μmol/L) whilst MAT(α1)2can be stimulated eight-fold by high SAMe levels (500 μmol/L)[33]. These differences between MATα1 and MATα2 are important to allow MATα1 to maintain a high SAMe production in the liver (produces 6-8 g/day)compared with MATα2, which does not contribute significantly to this SAMe pool[34].Indeed, by expressing MATα1 the liver is able to catabolize 50% of methionine intake via conversion to SAMe and allow an up to 10-fold rise in SAMe level following a methionine rich meal[35]. Consistent with this, cells that express MATα1 have much higher steady-state SAMe levels than cells that express MATα2[36]. When either major isoform of MATβ interacts with MATα2 they increase the kcat(turnover rate of an enzyme-substrate complex) of the MATαβ complexes[12,37,38]and increase the susceptibility to feedback inhibition by SAMe[37].
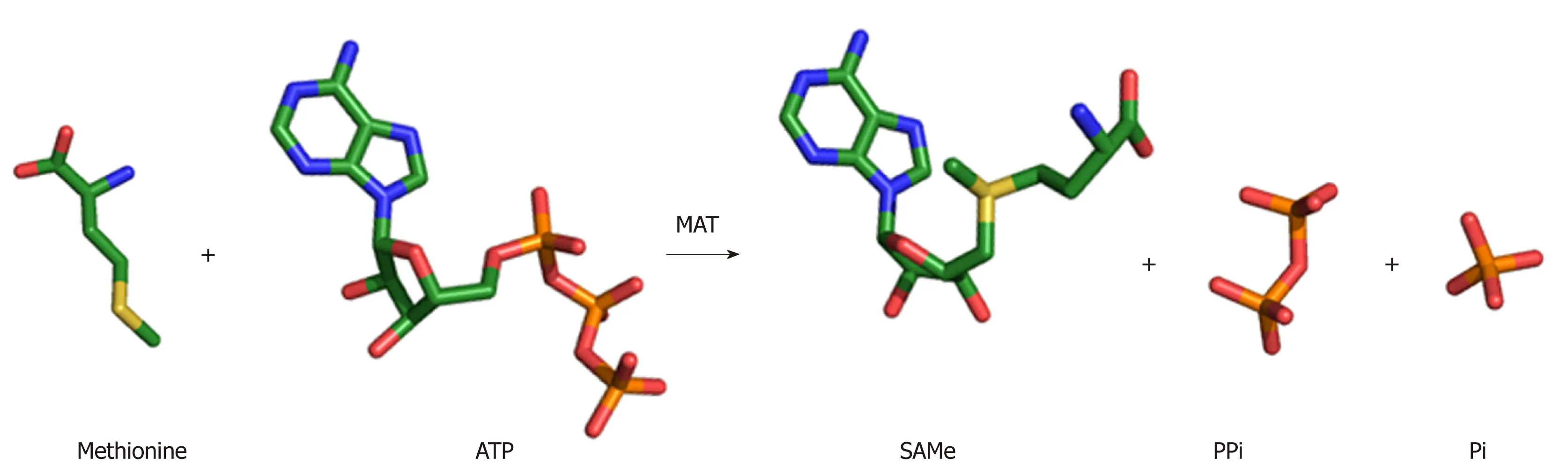
Figure 2 SAMe synthesis reaction. MAT enzyme catalyzes the biosynthesis of SAMe from the amino acid methionine and the energy molecule ATP. The sulphur atom of methionine attacks the C5' atom of ATP displacing the tripolyphosphate (PPPi) moiety to form SAMe. The PPPi is then hydrolyzed giving rise to pyrophosphate (PPi) and orthophosphate (Pi).
DYSREGULATION OF MATS IN LIVER DISEASE AND CANCER
Many studies have demonstrated that MAT proteins play important roles in chronic liver disease and hepatocarcinogenesis and a switch in their expression pattern is a frequent event in liver cancers. MAT1A, which is mainly expressed in the liver and maintains the differentiated state of hepatocytes, is downregulated in most cirrhotic patients[39], in patients with alcoholic hepatitis[40], during de-differentiation and in HCC[41,42]. Conversely, MAT2A and MAT2B, which are normally expressed only by non-parenchymal cells of the liver and extrahepatic tissues, are induced in HCC[14,42,43].This MAT1A to MAT2A/MAT2B switch contributes to reduced SAMe levels and is an important determinant of liver injury, fibrosis and liver cancer development in both rodents and humans[5].
MAT gene regulation and dysregulation in HCC
While MAT1A is a marker for normal differentiated liver, MAT2A and MAT2B are markers for rapid liver growth and de-differentiation. MAT2A and MAT2B are transcriptionally induced in human HCC, during rapid liver growth, dedifferentiation, and in response to ethanol feeding in rodents[44-46]. Reduced hepatic MAT activity has also been observed in cirrhotic patients, which explains why many cirrhotic patients have hypermethioninemia[39]. In human HCC, the MAT1A:MAT2A expression ratio has been inversely correlated with cell growth and genomic instability and directly correlated with HCC apoptosis and overall DNA methylation;a reduced ratio is a prognostic marker of more malignant and lower survival HCCs[47].MATs are regulated at transcriptional, post-transcriptional and post-translational levels by a complex network of mechanisms (Table 2). Many of these are dysregulated in HCC and participate to alter MATs expression.
MAT1A transcriptional and epigenetic control
The MAT1A promoter contains binding sites for multiple transcription factors,including hepatocyte nuclear factor (HNF), activator protein 1 (AP-1), CCAAT enhancer binding protein (C/EBP), c-MYC and glucocorticoids[48]. Some of these factors are determinants of liver-specific gene expression of MAT1A, such as HNF and C/EBP, with the latter also control MAT1A expression by promoter regulation[48,49].Prohibitin 1 (PHB1), which is highly expressed in normal hepatocytes and downregulated in most HCCs, positively regulates MAT1A mRNA levels[50]. Finally, c-MYC, MAFG and c-MAF, transcription factors that are overexpressed in human HCC and CCA, have been shown to bind to a repressive E-box element in the human MAT1A promoter to downregulate MAT1A transcription[8,9].
MAT1A expression is also regulated by DNA epigenetic modifications. Lower MAT1A expression in HCC has been associated with promoter hypermethylation and histone H4 deacetylation of its promoter[47,51]. Further investigation revealed a 750-base pair (bp) region upstream of the MAT1A transcriptional start site for these epigenetic modifications[51]. In HepG2 cells and cirrhotic human livers, hypermethylation of sites+10 and +88, relative to the transcriptional start site have been reported to also downregulate MAT1A transcription[52]. Low MAT1A mRNA levels and hypermethylation of both the MAT1A promoter and coding regions were also reported in patients with advanced NAFLD[53].

Table 2 Regulatory mechanisms of human MAT genes and proteins
MAT1A post-transcriptional control
Binding of AU-rich RNA binding factor (AUF1) to the 3'-untranslated region (UTR) of MAT1A mRNA negatively regulates its stability. There is an inverse correlation between AUF1 and Mat1a expression; de-differentiation of rat hepatocytes in culture increases the expression of AUF1, contributing to the fall in Mat1a mRNA levels,whereas during liver development AUF expression falls, which coincide with increased Mat1a expression; AUF1 is highly expressed in human HCC and its knockdown increased MAT1A mRNA levels[54].
MAT1A mRNA is also regulated by microRNAs (miRNAs) in HCC[55,56]. Injection of 2-acetylaminofluorene in rats resulted in preneoplastic liver lesions, induction of both miR-22 and miR-29b and inhibition of Mat1a mRNA expression[55]. MiR-485-3p, miR-495, and miR-664, which are increased in human HCC and responsible for the induction of LIN28B, an oncoprotein that is overexpressed in HCC and represses the tumor suppressor Let-7, have been shown to negatively regulate MAT1A. These specific miRNAs, through the downregulation of MAT1A, lowered nuclear SAMe levels, leading to hypomethylation of the LIN28B promoter region and increased LIN28B expression[56]. Inhibition of these miRNAs reduced tumor growth in vitro and in vivo by recovering MAT1A expression and inducing apoptosis[56].
MAT2A/MAT2B transcriptional and epigenetic control
MAT2A transcription is upregulated during liver growth and de-differentiation[57-59].Like MAT1A, MAT2A is also regulated by multiple transcription factors including c-MYB, E2F and specificity protein 1 (SP-1), all of which increase its promoter activity[57,59]. The MAT2A promoter can also be induced by tumor necrosis factor-α(TNF-α) via nuclear factor κβ (NF-κβ) and AP-1 elements in the promoter region[60].Transforming growth factor β1 (TGF-β1) also increases the activity of the MAT2A promoter via NF-κβ in hepatic stellate cells (HSC)[61]. Multiple PPAR response elements (PPRE) that bind nuclear receptors including peroxisome-proliferator activated receptors (PPAR) are present in the rat Mat2a promoter[62]. In normal liver PPARγ is a marker of HSC quiescence, whilst PPARβ is induced in activated HSCs during liver fibrogenesis[63,64]. Both PPARγ and PPARβ occupy the same site on the Mat2a promoter to cause opposite effects[62]. PPARγ negatively regulate Mat2a transcription but during HSC activation PPARγ expression and activity fall, allowing PPARβ to bind instead and induce Mat2a expression[62]. The MAT2A promoter is also regulated by methylation and acetylation and in human HCC, MAT2A promoter is hypomethylation and associate with higher histone H4 acetylation[58]. Expression of MAT2A is also induced in a hypoxic tumor environment because hypoxia-inducible factor-1α (HIF-1α) binds to a consensus sequence within the MAT2A promoter activating its expression in human hepatoma cells[65]. Finally, hepatitis B X protein(HBx) was shown to activate MAT2A gene transcription by facilitating NF-κB and CREB binding to the MAT2A promoter, explaining MAT2A induction in HBVassociated HCC[66].
Mechanism of MAT2B transcriptional regulation remains poorly characterized. In the human liver cancer cell line HepG2, TNF-α can upregulate MAT2B-V1 mRNA but not MAT2B-V2 through an AP-1 and NF-κβ dependent mechanisms[14]. Sirtuin 1(SIRT1), a NAD+-dependent deacetylase, can also induce the expression of MAT2B[67].MAT2B-V1 mRNA expression has also been found to be regulated by leptin in HepG2 cells by mechanisms that involve extracellular signal regulated kinase (ERK) and AKT[68].
MAT2A/MAT2B post-transcriptional control
The stability of MAT2A mRNA can be influenced by the human RNA-binding (HuR)protein and its methylated form, methyl-HuR[54]. The function of HuR depends on the methylated state of the protein with methyl-HuR destabilizing target mRNAs and HuR stabilizing them[54]. During hepatocyte de-differentiation as well as HCC, HuR is induced but there is a decline in methyl-HuR, which results in a higher HuR/methyl-HuR ratio. For MAT2A this causes increased binding of HuR to MAT2A mRNA stabilizing it in HCC and de-differentiated hepatocytes[54]. HuR has also been shown to stabilize MAT2B mRNA in a similar manner as MAT2A mRNA[67].
Drug-induced miRNAs including miR-21-3p have been shown to control MAT2A and MAT2B stability in HepG2 cells. Either treatment with the anticancer drug berberine, which induces miR-21-3p, or overexpression of miR-21-3p itself causes apoptosis and inhibition of growth by downregulating both MAT2A and MAT2B[69].Most recently miR-34a and miR-34b, tumor suppressor miRNAs that are downregulated in multiple cancers including HCC, were shown to directly target MAT2A mRNA and lower its expression[70].
The N6-methyladenosin (m6A) methyltransferase METTL16, has been shown to methylate the 3'UTR of MAT2A hairpins in HEK293T and HeLa cells[71,72]. In high SAMe conditions methylation of hairpin 1 (hp1) of the MAT2A 3'UTR promotes intron retention and nuclear degradation whilst low SAMe levels promote the enhanced binding of METTL16 to the hp1 of MAT2A 3'UTR leading to increased splicing and MAT2A translation[71,72]. Regulation of MAT2A by METTL16 has not been reported in HCC.
MAT proteins post-translational modifications
The activity and stability of MAT proteins can be altered by post-translational modifications including nitrosylation, phosphorylation and sumoylation. MATα1 has cysteine at position 121 (C120 in human), which lies within the flexible gating loop,can be both nitrosylated or oxidized leading to enzyme inactivation[73]. GSH, an antioxidant, and other thiol-reducing agents can prevent and reverse this inactivation[74]. MATα2 cannot be inhibited in this manner as glycine is at this position. Pho-sphorylation of MATα1 at threonine 342 (T341 in human) by protein kinase C was reported 25 years ago, and this modification does not alter the kinetic properties of this enzyme[75]. However, treatment with alkaline phosphatase to dephosphorylate T342 lowered the activity of both MAT(α1)2and MAT(α1)4in vitro[75].Whether this is true in vivo has not been examined.
MATα2 and MATβ have also been reported to be phosphorylated. During liver fibrosis and HSC activation, MATα2 and MATβ are phosphorylated via mitogen activated protein kinase/ERK kinase (MEK) and ERK, respectively, which leads to their stabilization[76]. Analysis revealed Y371/Y374 in MATα2 and T257/Y259 in MATβ to be the sites of phosphorylation and importantly, mutation of these residues inhibited HSC activation[76]. The use of in vitro kinase assays, gene silencing, and chemical inhibitors have shown that MEK could be responsible for the phosphorylation of MATα2, whilst MATβ may be modified by ERK[76].
Sumoylation is a post-translational modification that involves the conjugation of proteins with a small ubiquitin modifier (SUMO) that can lead to changes in the target protein's stability, localization, tertiary interaction and activity[77]. Attachment of SUMO-1 to a protein is achieved through the sole E2-conjugating enzyme, ubiquitinconjugating enzyme 9 (UBC9)[78]. The addition of SUMO-1 to a protein is generally associated with stability, and it has been shown that MATα2 has three SUMO-1 modifications at the lysine residues K340, K369 and K394, that increase the stability of MATα2 and also enhance its interaction with oncoproteins such as B-Cell CLL/lymphoma 2 (BCL-2)[77,79]. It has been shown that treatment with SAMe in liver cancer cells reduces the expression and activity of UBC9[80].
While in normal liver MATα2 acetylation at K81 by the E1A binding protein (P300)causes its ubiquitination and degradation, in liver disease a lack of acetylation stabilizes the protein, and this has been associated with HCC development that is attributed to the deacetylase HDAC3[81].
The effect of SAMe on MAT expression
While the expression of MATs can influence the steady-state levels of SAMe[36], SAMe level can, in return, influence MAT expression. During de-differentiation of primary hepatocytes in culture MAT1A expression falls while MAT2A expression is induced[82].This effect is due to a fall in SAMe level since it can be blocked by the addition of SAMe. Consistently, a fall in SAMe level (by restricting L-methionine in medium)leads to rapid induction of MAT2A expression that is blocked upon the addition of SAMe[58,83]. Treatment with SAMe results in higher methyl-HuR level leading to enhanced mRNA destabilization of MAT2A which may explain its negative effect on MAT2A expression[54]. SAMe also inhibits MAT2B expression at baseline and prevents leptin induced induction in hepatoma cells[68].
MAT proteins interactome
MAT proteins exhibit distinct interactomes in normal and diseased liver. All three MATs interact with a variety of proteins regulating their expression and contributing to liver injury and carcinogenesis.
MATα1 can act as a transcription co-factor by interacting with other E-box binding regulatory proteins. MATα1 can heterodimerize with MAX in HCC to repress E-boxdriven promoter activity, which results in the negative regulation of the transcription factors c-MYC, MAFG and c-MAF, and their oncogenic activity[9,50]. MATα1 has also been found to interact with CYP2E1 to negatively regulate its protein expression by inducing its proteasomal degradation[19]. Interestingly, MATα1 also interacts with p53 and DNA damage-regulated gene 1 (PDRG1) in hepatoma cells and in a mouse model of acute liver injury, which exhibited reduced total MATα1 expression but accumulation of nuclear MATα1[84]. PDRG1 is an oncogene that is upregulated in bladder, breast and colon cancer[85]. Interaction of PDGR1 with MATα1 in the nucleus resulted in a decrease in MAT activity and DNA hypomethylation[84]. The role of PDRG1 in HCC remains unknown.
MATα2 not only binds to and stabilizes BCL-2 protein, it also enhances BCL-2 transcription in liver and colon cancer cell lines by binding to its promoter[79]. MATβ is known to interact with HuR; when either of the MAT2B variants is overexpressed,cytosolic HuR content increases leading to higher mRNA levels of HuR targets such as cyclin D1 and cyclin A and proliferation[18]. MATβ also interacts with SIRT1, and resveratrol increases this interaction by stabilizing them[67]. MATβ also interacts with G-protein-coupled receptor kinase-interacting protein 1 (GIT1) to form a scaffold complex that interacts and activates all components of the RAS/RAF/MEK/ERK signaling pathway in liver cancer cells, promoting growth in vitro and in vivo[7].Interaction between MATβ and GIT1 appear to also stabilize MATβ[86]. Finally, both MATα2 and MATβ are often overexpressed in parallel in multiple cancers and part of the reason is that their interaction stabilizes both proteins[70].
CONSEQUENCES OF MAT GENES DYSREGULATION IN THE DEVELOPMENT OF HCC
MAT genes deregulation has been widely associated with alterations that contribute to liver disease and the development of HCC. MAT1A downregulation increases oxidative stress, progenitor cells expansion, genomic instability and other mechanisms implicated in tumorigenesis, whilst MAT2A and MAT2B, which are induced in HCC, confer growth and survival advantages to cancer cells (Reviewed in Lu 2012)[5]. In humans, the fall in MAT activity observed in cirrhotic patients is thought to contribute to the pathogenesis and progression of the disease as well as predisposition to HCC[39].
The Mat1a-KO mouse model
The Mat1a-KO mouse model has provided important insights into the mechanisms of how MAT1A might influence HCC development. This model is relevant to human liver disease since MAT1A expression is markedly reduced in the majority of cirrhotic patients[87]. Mice lacking Mat1a have markedly increased serum methionine levels and chronically reduced hepatic SAMe (70% lower) and GSH (40% lower) levels. By three months Mat1a-KO mice develop hepatic hyperplasia and are more susceptible to liver steatosis in response to a choline-deficient diet. By eight months, Mat1a-KO mice spontaneously develop NASH on a normal diet and by 18 to 20 months they develop HCC[88]. The livers of Mat1a-deficient mice also exhibit oxidative stress caused by low GSH levels, impaired mitochondrial function and increased expression of CYP2E1[34].CYP2E1 is the principal P-450 enzyme responsible for the metabolism of hepatotoxins such as alcohol, acetaminophen and CCl4in the liver and has a critical role in the generation of reactive oxygen species (ROS). Because of this, Mat1a-KO mice are more susceptible to CCl4-induced liver injury[34]. Mitochondrial dysfunction is another important mechanism that sensitizes Mat1a-KO mice to liver injury, which was mainly attributed to low levels of both the mitochondrial chaperone PHB1 and oxidative stress[34,89]. Our recent work adds loss of mitochondrial MATα1, an important regulator of mitochondrial function, as another mechanism[19].
Genomic instability
Genomic instability, which arises from the large number of genomic mutations,chromosomal aberrations, duplications, deletions and replication errors that cancer cells carry, is a characteristic of most cancers including HCC and is considered an early step in carcinogenesis[90,91]. Genomic instability may contribute to initiation of the malignant transformation, progression of the cancer and even resistance to therapy,influencing the overall prognosis of the cancer[92].
MAT1A regulates DNA methylation via SAMe and so MAT1A deficiency leads to DNA hypomethylation, which may contribute to genomic instability. Interestingly,alterations in the activity of MAT proteins and global DNA hypomethylation are prognostic markers for human HCC possibly through genomic instability[93]. These results suggest that early dysregulation of MAT proteins could influence the progression from preneoplastic lesions to cancer. In addition, the Apurinic/Apyrimidinic Endonuclease 1 (APEX1) protein, which participates in the base excision repair of premutagenic apurinic/apyrimidinic (AP) sites, the most frequent DNA lesion in cells, is markedly downregulated in Mat1a-KO livers. APEX1 protein level is reduced in de-differentiated hepatocytes with reduced Mat1a expression and low SAMe levels and is recovered by SAMe treatment[94]. Taken together, these findings demonstrate that hepatic SAMe depletion promotes genomic instability by different mechanisms contributing to malignant transformation.
Mitochondrial dysfunction
Along with oxidative stress, mitochondrial dysfunction represents an important trigger to hepatocarcinogenesis[95,96]. Although mitochondrial metabolism in malignant cells is controversial, deregulated cellular energetics associated with mitochondrial dysfunction are considered a common event in cancer[95]. As described above Mat1a-KO livers exhibit mitochondrial dysfunction from multiple mechanisms, including lower PHB1 expression, loss of mitochondrial MATα1 and increased expression of CYP2E1.
Decreased PHB1 expression
A proteomics study identified several mitochondrial proteins to be downregulated in Mat1a-KO mice from birth until the development of NASH[89]. PHB1 is a well-known mitochondrial chaperone that stabilizes newly synthesized mitochondrial proteins and maintains the organization and stability of mitochondrial nucleoids. PHB1 is essential for mitochondrial function and was found significantly downregulated in the livers of Mat1a-KO mice as compared to wild-type animals. In vitro experiment suggested low SAMe level promoted increased PHB1 degradation, as SAMe addition prevented the fall in PHB1 protein during culture[89]. However, a recent study showed PHB1 and MAT1A exert reciprocal positive regulation on each other at the mRNA level[50], adding another mechanism to low PHB1 expression in the Mat1a-KO liver.
The generation of a liver specific Phb1-KO mouse model confirmed that PHB1 hepatic deficiency predisposes to liver injury and malignant transformation. Liverspecific Phb1-KO mice have liver injury at a very young age, abnormal mitochondria and increased oxidative stress. Mice lacking Phb1 develop progressive fibrosis and multi-focal HCC by 8-10 mo of age[97]. Although HCC could have developed as a consequence of chronic inflammation, accumulating evidence support a tumor suppressor function of PHB1 in the liver. For instance, PHB1 silencing in murine nontransformed AML12 cells increased cyclin D1, H19, and IGF2 expression and enhanced E2F binding to the cyclin D1 promoter and proliferation[97]. PHB1 can cooperate with CCCTC-binding transcription factor (CTCF) to negatively regulate H19 and IGF2 expression, both of which are induced in HCC[98]. Reduced PHB1 expression has also been shown to induce IL-8 transcription by activating NF-κB and AP-1, resulting in enhanced IL-8 expression and release to promote tumorigenesis[99].Finally, PHB1 also acts as a negative regulator of WNT signaling, and its downregulation causes the induction of multiple WNT ligands and downstream activation of canonical WNT β-catenin signaling in murine liver and human HCC cells, in part through E2F[100].
It should be noted that the tumor suppressor role of PHB1 is highly controversial since PHB1 expression is increased in other cancers. PHB1 is also found in the nucleus, where it has been shown to interact with Rb and p53 among other proteins to bring about a change in transcriptional activities of E2F and p53[101]. Different subcellular localizations and post-translation modifications may explain the contradictory tumor regulatory activities of PHB1 in different cell types.
Mitochondrial MATα1
Recently MATα1 was shown to be present in the mitochondrial matrix in hepatocytes[19]. Mat1a-KO hepatocytes had reduced mitochondrial membrane potential and higher mitochondrial ROS, both of which were normalized when MAT1A was overexpressed. Oxygen consumption rate, ATP production and maximal and spare respiratory capacities were all reduced in Mat1a-deficient mitochondria,supporting the negative effect that MAT1A deficiency has on mitochondrial function.Another important finding that may contribute to mitochondrial dysfunction and liver injury in Mat1a-KO mice is the interaction of MATα1 and CYP2E1 in the mitochondria. Mat1a deficiency leads to higher CYP2E1 mitochondrial levels.Mitochondrial CYP2E1 also regulates the production of ROS and is known to contribute to liver injury and mitochondrial dysfunction[102,103]. MATα1 negatively regulates CYP2E1 expression at mRNA and protein levels, with the latter being the dominant mechanism that involves methylation of CYP2E1 R379, promoting its proteasomal degradation[19].
MATα1 was found to also interact with important mitochondrial proteins including several subunits of the electron transport chain complexes, which raises the possibility that MATα1 could be regulating mitochondrial function in multiple ways. These findings highlight a critical role of MATα1 in regulating mitochondrial function and could provide a novel target for the treatment of different liver diseases where mitochondrial dysfunction plays a key role. Taken together, reduced mitochondrial MATα1 could play a key role in the mitochondrial dysfunction that is often observed in HCC[104].
Cancer stem cells
Cancer stem cells, also known as tumorinitiating cells, are known to play a central role in tumor development, metastasis and recurrence and are considered key therapeutic target for cancer treatment. Hepatic oval cells are the cancer stem cells of the liver and important contributors of hepatocarcinogenesis[105]. They are quiescent in normal adult liver and low in number and expand during severe and prolonged injury as seen in various models of experimental carcinogenesis[106].
Methyl-deficient diets have been used to induce oval cell proliferation and HCC formation in susceptible models such as p53-/-mice[107]. Mat1a-KO livers contain increased populations of liver cancer stem cells (or CD133+/CD49f+oval cells), as they age[108]. These cells have increased expression of several oncogenes and are tumorigenic in vivo. Interestingly, Mat1a-KO's cancer stem cells show increased MAPK signaling with enhanced ERK activity, like Mat1a-KO's hepatocytes, and increased oncogenic signaling (K-Ras and Survivin)[108]. Constitutive ERK activation makes these cells resistant to the apoptotic effect of TGF-β, a well-known growth inhibitor in hepatocytes[109,110]. How SAMe deficiency allows expansion of oval cells remains unknown.
Dysregulated pathways
Increasing evidence indicates that the deregulation of various signaling pathways progressively increase with HCC progression and has a prognostic value. The MAT1A to MAT2A/MAT2B switch is also associated with activation of multiple signaling pathways including RAS/ERK[76,86,111], IκB kinase (IKK)/NF-kB[14,60], Phosphoinositide 3-kinase (PI3K)/AKT[68,112,113]and Liver kinase B1 (LKB1)/AMPK-activated protein kinase (AMPK)[114]. Increased sumoylation[80]and c-MYC expression[8], which are wellknown contributors of hepatocarcinogenesis, have also been associated with reduced hepatic SAMe levels.
ERK signaling
ERK signaling is tightly regulated in normal cells but uncontrollably active in cancer cells, being one of the several growth signals associated with highly malignant HCCs[115,116]. ERK activity is regulated by the dual-specificity MAPK phosphatase(DUSP1). DUSP1 is a member of a family of dual-specificity MAPK phosphatases that dephosphorylates both serine/threonine and tyrosine residues[117]. Interestingly, there is a reciprocal regulation between DUSP1 and ERK[111]. Transient activation of ERK leads to catalytic activation DUSP1, which in turn inhibits ERK activity by dephosphorylation[118]. DUSP1 feedback inhibits ERK and this activity of DUSP1 is crucial for the regulation of ERK activity in liver cells. However, prolonged ERK activation induces the phosphorylation of DUSP1 at the Ser296 residue rendering the DUSP1 protein susceptible to proteasomal degradation[119]. In human HCC, DUSP1 expression is negatively correlated with proliferation and microvessel density and positively with survival[118].
Hepatic DUSP1 expression is decreased in Mat1a-KO mice both at the mRNA and protein levels, being more pronounced at the protein level and was normalized after SAMe treatment[111]. SAMe increased DUSP1 mRNA level by enhancing p53 binding to its consensus element in the DUSP1 promoter, which is known to activate DUSP1 transcriptionally[111]. Increased binding of p53 to the DUSP1 promoter in SAMe fed mice was due to the fact that SAMe stabilizes APEX1, which is a known transactivator of p53[94]. DUSP1 lower protein level was attributed to its faster degradation due to increased proteasomal activity in Mat1a-KO mice. SAMe treatment normalized proteasomal activity, increasing DUSP1 protein level and normalized ERK activity in Mat1a-KO mice[111]. These findings suggest that SAMe deficiency leads to uncontrolled ERK activation due at least in part to decreased DUSP1 expression during HCC development. ERK signaling was also found to be regulated by MAT2B since its knockdown inhibited the activation of MAPK/ERK pathway induced by leptin in liver cancer cell lines[68]. Finally, as mentioned earlier, the MATβ-GIT1 complex efficiently binds to MEK and ERK leading to their activation. Consistent with this,overexpression of MAT2B or GIT1 in the HCC cell line Huh7 enhanced tumor growth and metastasis in a mouse orthotopic HCC model[86].
LKB1-AMPK signaling
Even though LKB1 is considered a tumor suppressor in a variety of cancers[120], its role in liver carcinogenesis remains controversial given the fact that both reduced and increased LKB1 levels have been reported in human HCC correlating with prognosis[121-124]. In recent years, several publications have supported the oncogenic role of LKB1 in liver cancer[122-125].
LKB1 phosphorylates and activates AMPK, a central metabolic sensor, to control cell growth in response to environmental nutrient changes. Indeed, the LKB1/AMPK signaling pathway has tumor suppressor activity since it serves as a metabolic checkpoint arresting cell growth in conditions of nutrient deprivation or low intracellular ATP levels[126]. In the liver the activation of LKB1 and AMPK leads to the production of nitric oxide (NO) and is required for hepatocyte proliferation, as seen in regenerating livers after partial hepatectomy and hepatocyte growth factor (HGF)treated rat hepatocytes[114]. Interestingly, early during liver regeneration hepatic SAMe level falls and exogenous SAMe inhibits regeneration[127]. The thought is that SAMe level needs to fall in order to release the inhibitory tone it exerts on mitogenic pathways. Although Mat1a-KO mice have higher basal proliferation, they exhibit impaired liver regeneration because hepatic SAMe level remained unchanged[127].Consistent with this, in the presence of SAMe, protein phosphatase 2A (PP2A)interacts with AMPK, leading to its dephosphorylation and inactivation. One explanation for the fall in hepatic SAMe level early in liver regeneration is due to increased NO formation, which can inactivate MATα1[128,129].
Activation of the LKB1/AMPK pathway may contribute to hepatocarcinogenesis through other mechanisms as well. Increased LKB1 and AMPK activity results in nuclear to cytoplasmic HuR translocation and the subsequent stabilization of several cyclin mRNAs enhancing cell proliferation[130]. LKB1/AMPK activation is also required for survival of HCC cells derived from Mat1a-KO livers, named SAMe-D[114].LKB1 can regulate AKT-mediated cell survival independent of PI3K, AMPK, and mTOR2 (mammalian target of rapamycin complex). LKB1 is hyperactivated in SAMe-D cells and can control survival through the phosphorylation and cytoplasmic retention of p53. In normal cells, p53 expression is maintained at a low level and is inactivated by Mdm2-mediated nuclear to cytosol transportation and proteasomal degradation. In response to oncogenic insults, p53 translocates to the nucleus to exert its tumor suppressor activity and activate pathways associated with DNA repair, cell cycle arrest and apoptosis. Notably, HuR nucleocytoplasmic shuttling also stabilizes p53 mRNA. Supporting these findings, increased cytoplasmic staining of p53 and phospho-LKB1 were found in the Mat1a-KO livers and in livers from human HCC derived from both NASH and alcoholic steatohepatitis[131]. Figure 3 summarizes the mechanisms of HCC development in the Mat1a-KO mouse model.
MAT DYSREGULATION IN CCA
CCA is, after HCC, the second most common primary hepatic malignancy and like HCC, its development involves MAT genes deregulation. MAT1A is highly expressed also in normal bile duct epithelial cells and is repressed during chronic cholestasis and in murine and human CCA[8]. There are common mechanisms of MAT genes deregulation between HCC and CCA. For example, hypermethylation of the MAT1A promoter has also been observed in CCA. The transcription factors c-MYC, MAFG and c-MAF, which are all induced both in HCC and CCA, also negatively regulate MAT1A transcription in CCA by binding to its repressor E-box promoter region[8].PHB1, which is also downregulated in most human CCAs, positively regulates MAT1A while suppressing c-MYC, MAFG, and c-MAF expression in mice[9,50].Consistently, reduced PHB1 expression predisposes to the development of cholestasis-induced CCA[8,50]. Figure 4 summarizes changes in MATs and SAMe in HCC and CCA as compared to normal liver.
TARGETING MATS IN LIVER CANCER
Restoration of SAMe levels
The Mat1a-KO mouse model demonstrates that the switch from Mat1a to Mat2a leads to hepatic SAMe deficiency and hypermethioninemia[88]. Patients with human liver cirrhosis[39], alcoholic hepatitis[40], advanced NAFLD[53], as well as HCC[42]all have lower MAT1A mRNA level. The restoration of hepatic SAMe level appears to be an obvious therapy to overcome the fall in SAMe level due to the MAT1A/MAT2A switch. There have been very few human trials that examined SAMe in chronic liver disease. One study showed that SAMe treatment (1,200 mg orally per day in three divided doses) for two years improved the survival or delayed liver transplantation in patients who had alcoholic liver cirrhosis with less advanced liver disease (Child's class A and B)[132]. However, this was a post-hoc analysis, so it remains to be confirmed. Another study in patients with hepatitis B-related advanced-stage (stages B-C) HCC who received 1000 mg of SAMe two hours before surgery intravenously and continued for five consecutive postoperative days showed a reduction of alanine aminotransferase and aspartate transaminase levels, delayed recurrence and a greater 24-mo survival rate as well as a lower risk of complications[133]. However, analysis by the Cochrane Hepato-Biliary group using a total of 330 alcoholic liver disease patients treated with SAMe in 8 clinical trials did not confirm the beneficial effects of SAMe in human liver cirrhosis although they were small with variable quality, which resulted in the meta-analysis failing to show a significant benefit of SAMe treatment[134]. At the present time the verdict on the effectiveness of SAMe in chronic liver disease is still out and its utility in chemoprevention of HCC remains to be studied.
In CCl4-induced liver injury SAMe treatment prevented the activation of HSCs by inhibiting the collagen promoter as well as downregulating TGF-β-induced extracellular matrix protein, α-smooth muscle actin (α-SMA)[135]. It has also been demonstrated that SAMe is selectively pro-apoptotic in liver cancer cells but antiapoptotic in normal hepatocytes[136,137]. This property makes SAMe particularly attractive as a chemopreventive agent. Indeed, SAMe can chemoprevent against HCC in several preclinical models[5]. However, SAMe was ineffective at treating established tumors in an orthotopic HCC model[138]. Investigation into this revealed that buildup of SAMe in the liver was prevented by the compensatory induction of glycine-N methyl transferase (GNMT), an important enzyme abundantly expressed in the liver that is responsible for catabolizing SAMe and keeping SAMe level within a tight range[5,138]. Although acute pharmacologic SAMe treatment can transiently increase liver SAMe level by 10-fold, prolong SAMe treatment induced GNMT expression so that SAMe level increased only 30%, which is insufficient to exert a pro-apoptotic effect[138]. However, in human cirrhosis and HCC GNMT express is often downregulated[139], so the efficacy of SAMe for HCC treatment in human warrants further investigation. At present, there are two SAMe ongoing clinical trials in HCC treatment. One (NCT03178929) is designed to investigate SAMe treatment (2000 mg,P.O.) in HCC patients (Barcelona Clinic Liver Cancer 0/A) after radical treatment,where tumor recurrence is the endpoint. The other (NCT02586285) aims to study the SAMe treatment (500-1000 mg, iv, per day) in HCC patients after curative treatment.Although targeting SAMe has been the central focus for treatment, therapies that target the MAT proteins themselves could also offer potential benefit to which some of these are described below.
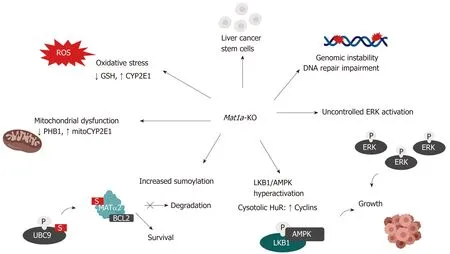
Figure 3 Mechanisms of hepatocellular carcinoma development in the Mat1a knockout mouse. Multiple mechanisms are known to influence hepatocellular carcinoma development in the Mat1a-KO mouse. These include: oxidative stress due to lower GSH levels and higher CYP2E1 expression; mitochondrial dysfunction due to reduced PHB1 and increased mitochondrial CYP2E1 levels; increased sumoylation, stabilizing MATα2, which acts as a transcription factor to enhance BCL-2 transcription as well as directly interacting with BCL-2 leading to its stabilization; enhanced activation of the LKB1/AMPK pathway which leads to the cytoplasmic translocation of HuR from nucleus and subsequent stabilization of cyclins; aberrant activation of ERK which promotes uncontrolled cell growth; enhanced genomic instability due to DNA hypomethylation and impaired DNA repair machinery and increased number of liver cancer stem cells with tumorigenic potential.
Restoring the expression of endogenous MAT1A
Overexpressing MAT1A in the liver cancer cell line Huh7 resulted in a stable increase in SAMe levels, an induction in tumor suppressor genes, downregulation of angiogenesis genes, reduced cell growth and increased apoptosis in vitro and in vivo[140]. This is a proof of principle that raising MAT1A expression in liver cancer could be an effective treatment strategy. While delivering MAT1A gene therapy to HCC cells is not a suitable approach, targeting miRNAs that downregulate MAT1A expression might be feasible. MiR-485-3p, miR-495, and miR-664 are three miRNAs that are increased in human HCC that directly target MAT1A at the 3'UTR[56].Treatment with siRNA against any of the miRNAs resulted in higher MAT1A expression, reduced HCC growth in an orthotopic HCC model[56]. Higher SAMe level as a result can also downregulate MAT2A/MAT2B to slow HCC growth. Taken together, raising endogenous MAT1A expression and hence SAMe level in the HCC cells is an attractive treatment approach that deserves further study.
Targeting of MAT proteins
Therapeutic agents to target MATα2 have been proposed for many years. In the 1970s methionine analogs were suggested to act as substrate-competitive inhibitors and proposed as chemotherapeutic agents[141]. Stilbene derivatives (FIDAS agents) have been proposed as inhibitors of MATα2, but these compounds are unlikely to specifically inhibit MATα2 because of their ability to bind multiple proteins targets[142].Using molecules that directly target the active site of MATα2 may be problematic as this protein is expressed in most extrahepatic cells which require MATα2 as the SAMe generator. In addition, the high level of sequence and structural identity between MATα1 and MATα2 makes it difficult to design active site molecules that would only target one these proteins. One strategy is to target the interaction of the MATα2β complexes as these complexes provide a proliferative advantage to HCC cells by lowering steady state SAMe level, stabilizing each other as well as activating several mitogenic pathways[7,43,68,86]. A novel allosteric inhibitor has been shown to bind to the interacting site of these proteins, which demonstrated promise in lung cancer cells[13].It remains to be examined in liver cancer models.
Targeting a posttranslational modification could also be a good strategy to only target MAT proteins that are contributing to the pathogenesis of HCC. Designing a molecule for a specific modification on MATα1, MATα2, or MATβ may be possible to stabilizes MATα1 whilst destabilizing MATα2 and MATβ. For instance, sumoylation on MATα2 facilitated BCL-2 induction in HCC, and induced chemoresistance in HCC cells[79]. Using small molecules to block the addition of SUMO to MATα2 would lower its expression as well as that of BCL-2. Similarly, MATα2 and MATβ are both stabilized by phosphorylation at specific residues which facilitated HSC activation[76].HSC activation is an important mediator of liver fibrosis, and therefore blocking the site-specific phosphorylation of these MAT proteins could be a novel treatment for liver fibrosis. As mentioned before, MATβ has been implicated to promote cell survival in HCC due to its interaction with the MEK/ERK/MAPK pathway[68,86]. The discovery that MATβ interacted with GIT1 and caused the amplification of RASmediated MAPK activation provides another novel target where a molecule could block this protein-protein interaction to inhibit MAPK signaling and cell proliferation[7].
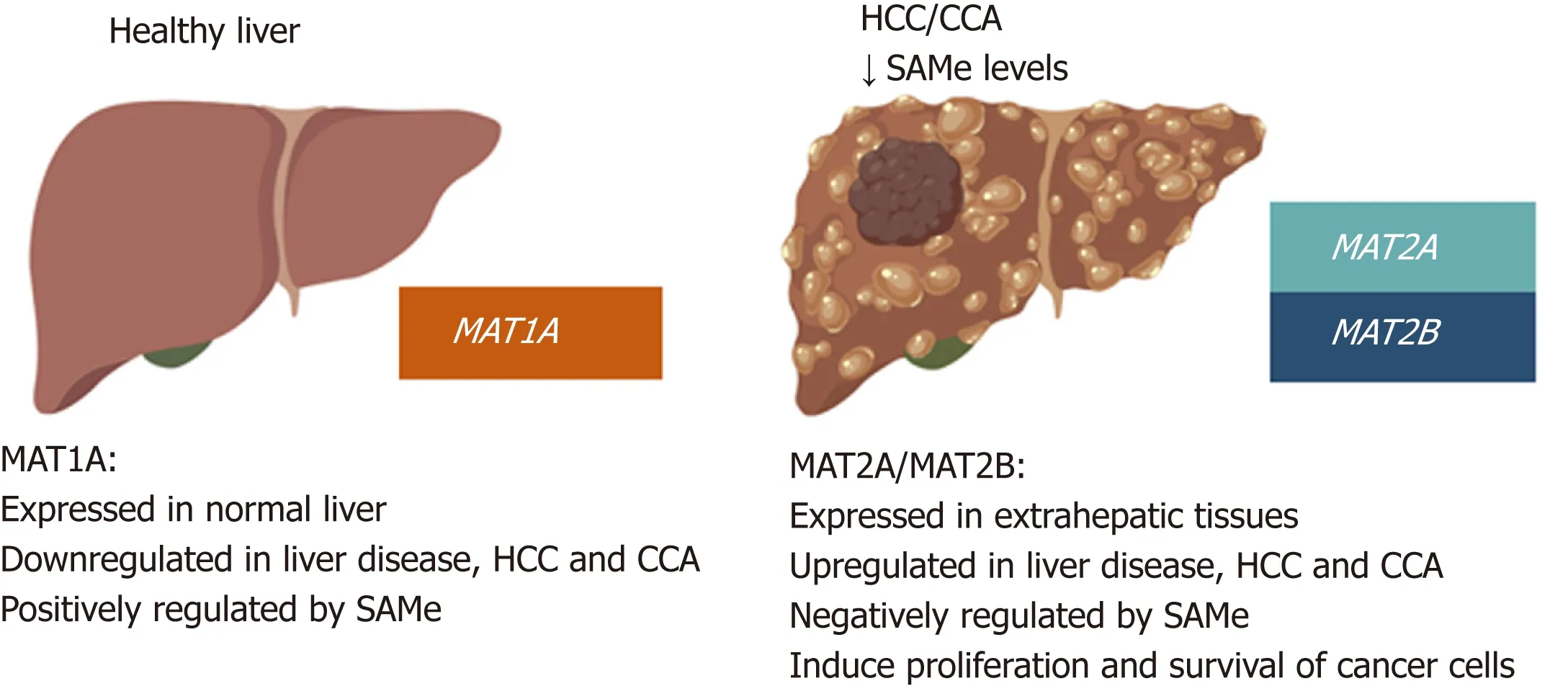
Figure 4 MAT genes expression pattern in normal liver and liver cancer.MAT1A is mainly expressed in normal liver by hepatocytes and bile duct epithelial cells; whereas MAT2A and MAT2B are expressed in extrahepatic tissues and by the non-parenchymal cells in the liver. During liver disease and malignant transformation there is a switch from MAT1A to MAT2A/MAT2B, being MAT2A and MAT2B the predominant MAT genes expressed in HCC and CCA.MAT1A maintains the differentiated state of the liver whilst MAT2A and MAT2B give proliferative and survival advantages to cancer cells. SAMe positively regulates MAT1A and negatively regulates MAT2A and MAT2B as well as feedback inhibits MATII. Accordingly, steady state SAMe levels are lower in patients with chronic liver disease and liver cancer due to the switch from MAT1A to MAT2A/MAT2B.
MATs as potential molecular markers in liver cancers
Epigenetic alterations including DNA methylation are recognized as a major characteristic in HCC and may serve as diagnostic and prognostic biomarkers[143].MATα1 to MATα2 switch and low SAMe level are associated with HCC development and strongly predict patients' survival[47,144]. High MAT1A expression in human HCC is negatively correlated to tumor size (> 5 cm), whilst up-regulation of MAT2A is positively correlated to serum alpha-fetoprotein level and one-year recurrence after hepatectomy[145]. Moreover, the MATα1 to MATα2 switch may promote HCC invasion and metastasis and lower patient recurrence-free survival[146]. All these suggest MATs may serve as diagnostic and prognostic molecular markers in HCC and likely CCA as well.
CONCLUSION
Taken together, accumulating evidence demonstrates the importance of MAT genes in liver tumorigenesis. Part of the mechanism is related to a lowering in the steady state SAMe level. However, all three MATs are found in the nucleus where they regulate gene expression via epigenetics as well as bona fide transcription factor and cofactors.They also have distinct interactomes and affect a myriad of signaling pathways. SAMe is an attractive chemopreventive and possibly therapeutic agent in liver cancer and its efficacy in both warrant investigation. Small molecules that inhibit interactions between MATβ-GIT1 or MATα2-β, both of which provide proliferative advantages to liver cancer cells, and targeting posttranslational modifications that can increase MAT1A and/or lower MAT2A/2B are all exciting future directions for translating the knowledge gained from understanding mechanisms of MAT dysregulations to therapy in liver cancer.
杂志排行

World Journal of Gastroenterology的其它文章
- lleal-anal pouches: A review of its history, indications, and complications
- Common features between neoplastic and preneoplastic lesions of the biliary tract and the pancreas
- Treatment of hepatocellular carcinoma in patients with portal vein tumor thrombosis: Beyond the known frontiers
- Systemic inflammation in colorectal cancer: Underlying factors,effects, and prognostic significance
- Overview and comparison of guidelines for management of pancreatic cystic neoplasms
- Gastrointestinal motility and absorptive disorders in patients with inflammatory bowel diseases: Prevalence, diagnosis and treatment