Zr,Nb,V在a-Fe(C)中的占位、电子结构及键合作用的第一性原理研究*
2019-08-27刘飞文志鹏
刘飞 文志鹏
1)(天津理工大学中环信息学院机械工程系 天津 300380)
2)(四川省宜宾市普什智能科技有限公司,宜宾 644000)
1 引 言
钢的微合金化技术是改善钢铁材料力学性能的重要方法[1−4].一方面,通过添加微量合金元素,形成a-Fe(C)合金固溶体提高强度; 另一方面,合金元素会与钢中的C,N等元素形成沉淀第二相,抑制奥氏体长大,细化晶粒达到提高塑性、韧性的目的.目前,国内外学者对Ti,Cr,Nb,Mo等元素对钢合金化作用的机理进行了理论及试验研究[5−9],并通过研究微合金碳氮化物的析出动力学来探讨析出相对奥氏体晶粒细化的作用.Zr,Nb,V均是强碳化物形成元素,这三种元素的微合金化能够对钢的力学性能产生一定影响.习天辉等[10]研究了Zr含量对微合金钢组织和性能的影响.唐兴昌等[2]对Nb,Ti微合金化低碳贝氏体高强钢组织性能及再结晶行为进行了研究.高绪涛等[11]对Nb,V,N微合金化热轧TRIP钢的动态连续冷却相变进行了研究.惠亚军等[12]对650 MPa级V-N微合金化汽车大梁钢强化机理进行了研究.近年来,相关学者对Zr,Nb,V元素细化晶粒及Zr-Ti,Nb-V复合微合金化的影响做了较多的研究工作[10,13].Zr易与C,N形成ZrC,ZrN,对微合金钢的力学性能产生了重大的影响,Zr也可以与过渡族金属Nb,Ti,V等相结合,通过控制晶粒尺寸提高屈服强度[14,15].He和Baker[14]研究发现,Zr可以起到细化奥氏体晶粒的作用.相关研究表明[16,17],向钢中添加部分Nb取代V,可以在高温下利用NbC抑制奥氏体晶粒的长大.然而,上述对Zr,Nb,V微合金化钢力学性能的影响仅基于实验及组织分析,三种元素对钢合金化的作用机理尚缺乏从微观电子结构的进一步讨论.虽然Zr,Nb,V均是强碳化物形成元素,但由于其与C原子的亲和力不同,因此,Zr,Nb,V三种元素对钢微合金化的作用机理可能存在一定差异.目前对相关研究尚无报导.
铁素体(a-Fe(C))是大部分钢材料的基体组织,在很大程度上决定着钢铁材料的使用性能.因此,研究合金元素对铁素体性能的影响对合金钢的设计及应用具有重要的理论及实际指导意义.近年来用第一性原理计算方法处理非均匀相互作用多粒子体系问题已经在计算凝聚态物理、计算材料科学等诸多领域取得了广泛应用.文献[18,19]采用第一性原理计算方法分析了Mn,Si,Ti,N,C等合金原子在a-Fe合金中的键合性质及合金化效应,但采用第一性原理方法从电子结构角度探讨合金钢中Zr,Nb,V等原子与a-Fe(C)相互作用的文章尚不多见.
本文基于密度泛函理论,采用第一性原理的计算方法研究了Zr,Nb,V元素对a-Fe(C)的合金化效应,从电子结构等方面解释了Zr,Nb,V合金原子与a-Fe(C)的微观作用机理,以期揭示铁素体固溶强化作用的本质,为合金设计的理论发展积累数据和方法.
2 晶体结构与计算方法
a-Fe的晶体结构中,空间点群为Im-3m,晶格常数采用a=b=c=2.866 Å.论文建立了1×1×2的a-Fe超晶胞模型,并在八面体间隙掺杂C原子,作为a-Fe(C)的初始结构模型.在此基础上,采用合金原子M(M=Zr,Nb,V)分别替代a-Fe(C)结构模型中体心以及顶角的Fe原子,获得了Zr,Nb,V原子在a-Fe(C)-M中不同占位的置换固溶体模型[20],如图1所示.其中紫色球表示Fe原子,灰色球表示C原子,蓝色球表示合金原子M.
材料结构和电子性质的计算基于DFT理论,采用CASTEP(Cambridge Serial Total Energy Package)软件[21,22].计算中,选取广义梯度近似(GGA)[23−25]框架下的PBE泛函形式作为交换关联函数,自洽求解了Kohn-Sham方程.采用超软赝势[26]描述价电子与离子实之间的相互作用,倒易空间中平面波计算的最大截止能量[27]为400.0 eV,自洽循环收敛精度为1.0 × 10–6eV/atom,自洽运算总能量收敛为1.0 × 10–5eV/atom,力收敛为0.3 eV/nm,公差偏移<2.0 × 10–4nm,应力偏差<0.05 GPa.布里渊区K矢量选取为6 × 6 × 3,自洽迭代的最大次数为200次.电子总能自洽用Pulay密度混合算法.计算力学性能时,在结构优化的基础上,第一布里渊区K矢量选取为9 × 9 × 4.
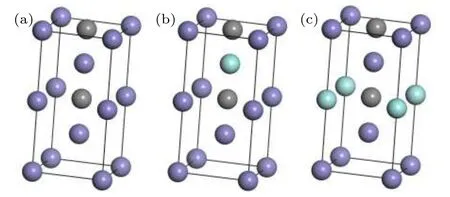
图1 Zr,Nb,V原子在a-Fe(C)-M中不同占位的置换固溶体模型Fig.1.Solid solution models of Zr,Nb and V atoms with different occupancy in a-Fe(C)-M.
3 结果与分析
3.1 合金原子固溶占位分析
Zr,Nb,V原子在a-Fe(C)中的固溶占位情况将会使合金铁素体发生不同程度的晶格畸变,从而影响其力学性能,力学性能将在本文3.4节进行讨论.合金原子在a-Fe(C)中有两种可能的固溶位置,即置换体心的Fe原子(如图1(b))和置换顶角的Fe原子(如图1(c)).合金原子的优先占位情况可以通过超晶胞固溶前后体积变化率e和超晶胞总能Etot来反映,其中e=(V–V0)/V0,V为a-Fe(C)-M超晶胞体积,V0为a-Fe(C)超晶胞体积.计算结果如表1所列.
从表1可以看出,Zr,Nb原子取代体心位置的Fe原子时,晶胞体积变化率较小,晶胞总能量相对较低; V原子取代顶角位置的Fe原子时,晶胞体积变化率较小,晶胞总能量相对较低.说明形成合金固溶体时,Zr,Nb原子优先取代a-Fe(C)晶胞中体心位置的Fe原子,而V原子优先取代a-Fe(C)晶胞中顶角位置的Fe原子形成a-Fe(C)-M合金铁素体.值得注意的是,Zr,Nb原子取代体心位置的Fe原子时,晶胞的体积变化率要大于V原子取代顶角位置的Fe原子.晶胞体积变化率及晶胞总能越小,说明晶胞越稳定,合金原子越容易固溶.因此得出,虽然Zr,Nb原子优先取代体心位置的Fe原子,但相比较而言,Zr,Nb原子比V原子的固溶要困难.
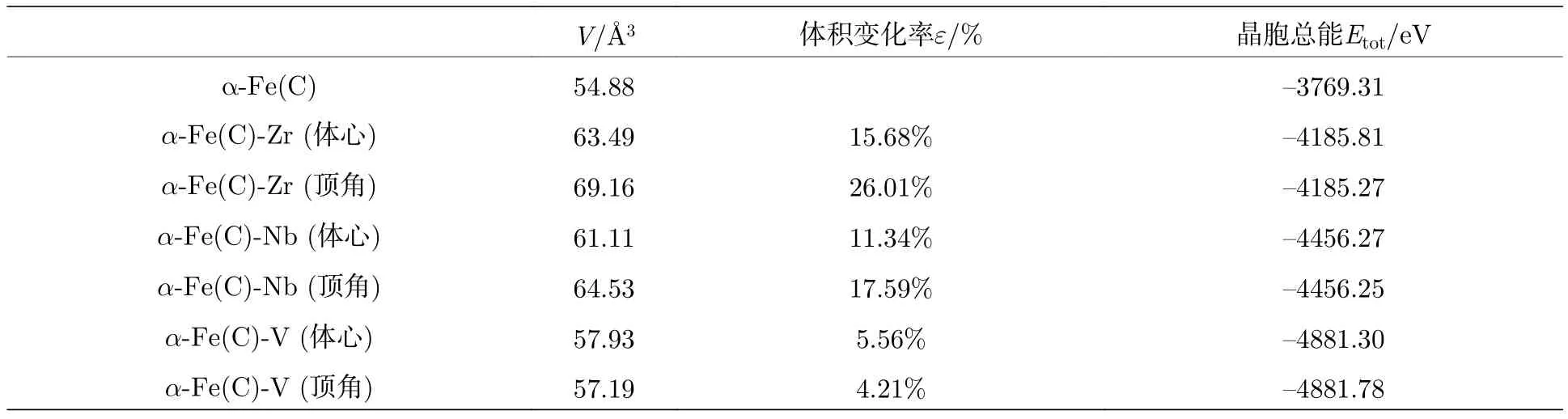
表1 a-Fe(C)-M晶胞体积变化率和晶胞总能Table 1.Cell volume change rate and total cell energy of a-Fe(C)-M.
3.2 结合能分析
合金铁素体的力学性能、结构稳定性与a-Fe(C)-M晶胞的结合能密切相关.晶胞结合能越大,原子的结合力越大,则需要更高的能量才能使键断裂,晶胞结构也就更稳定,同时破坏该原子的化学键也需要更大的能量.结合能按(1)式进行计算.

其中E0为晶胞的结合能,n为晶胞内的原子数,En为该晶胞内原子处于自由状态时的总能量,Etot为该晶胞的总能(计算结果列于表1).本文计算了Fe,C,Zr,Nb,V自由原子的能量,分别为–861.33,–148.32,–1278.94,–1548.98和–1973.43 eV,其中Fe,C的原子能量与文献[20]计算的结果基本一致(–859.82,–145.88 eV),说明了该计算方法的准确性.结合表1,本文计算了a-Fe(C)-M的结合能,结果如表2所列.

表2 a-Fe(C)-M晶胞结合能Table 2.Bonding energy of a-Fe(C)-M.
可以看出,Zr固溶后,晶胞的结合能降低了0.14 eV,Nb固溶后,晶胞的结合能降低了0.07 eV,说明Zr,Nb原子在一定程度上破坏了a-Fe(C)晶胞的稳定性,Zr相比于Nb原子更难固溶,这也与前节分析的结果一致.V固溶后,晶胞的结合能增加了0.13 eV,说明V原子的固溶增加了晶胞的稳定性,对于稳定铁素体具有一定意义.因此可以推测,Zr,Nb极有可能优先形成碳化物而非固溶于a-Fe(C)晶胞.同时,Zr比Nb更难固溶,也能在一定程度上说明Zr形成碳化物的倾向要强于Nb.这与合金钢理论中,Zr,Nb属于强碳化物形成元素,且Zr与C的亲和力要大于Nb这个结果完全一致.相比之下,V能够在一定程度上固溶a-Fe(C)晶胞,改善铁素体的力学性能.Zr,Nb,V对结合能的影响一方面与原子在a-Fe(C)中的占位有关,另一方面则需要从电子结构进行分析,从成键的角度来进行合理解释.
3.3 电子成键分析
态密度是分析电子是否成键的主要方法.本文计算了a-Fe(C)-M超晶胞的态密度(DOS)和分波态密度(PDOS),结果如图2所示,图2能较好地反映出Zr,Nb,V合金原子与a-Fe(C)晶胞中的Fe,C原子的成键情况.各原子轨道出现轨道分布能量的重叠,是原子参与成键的主要标志.
从图2(a)中可以看出,a-Fe(C)-Zr晶胞中,在不同能量范围内Fe,C,Zr原子不同轨道发生峰的重叠,表明Fe,C,Zr原子参与成键,提供成键电子.在–5.3—–3.3 eV处,PDOS图中Fe3d和C2p轨道出现轨道分布能量的重叠,Fe与C成键;在1.1—3.5 eV处,C2p和Zr4d轨道出现轨道分布能量的重叠,C与Zr成键.同理,从图2(b)中可以看出,a-Fe(C)-Nb晶胞中,在–5.3—–3.4 eV处,PDOS图中Fe3d和C2p轨道出现轨道分布能量的重叠,Fe与C成键; 在2.5—5.4 eV处,C2p和Nb4d轨道出现轨道分布能量的重叠,C与Nb成键.从图2(c)中可以看出,a-Fe(C)-V晶胞中,总态密度上下自旋出现明显不对称情况,说明V原子的固溶使晶胞体系出现了磁性.在0.3—1.9 eV处,PDOS图中Fe3d和V3d轨道出现轨道分布能量的重叠,Fe与V成键; 在1.9—3.3 eV处,V3d和C2p轨道出现轨道分布能量的重叠,V与C成键;在4.8—8.6 eV处,Fe3p和C2p轨道出现轨道分布能量的重叠,Fe与C成键; V原子虽然在–65.2—–63.7 eV及–39.4—–37.5 eV区间内提供成键电子,但Fe和C在这两个区间的态密度几乎为零; C原子虽然在–14.5—–10.2 eV区间内提供成键电子,但Fe和V在这两个区间的态密度几乎为零.因此,在a-Fe(C)-V晶胞中,三种原子都提供成键电子,主要形成Fe—C键、Fe—V键及V—C键,参与成键的主要是Fe3p,C2p,Fe3d及V3d轨道.
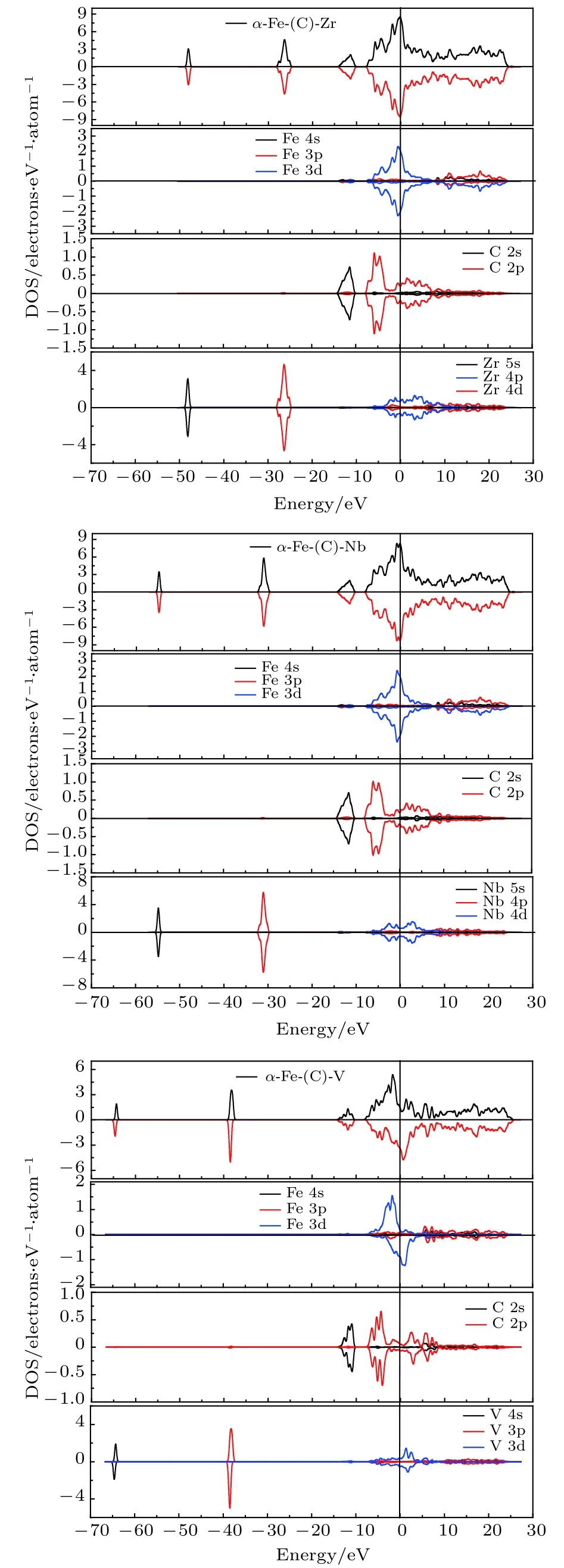
图2 a-Fe(C)-M合金的态密度和分波态密度图Fig.2.DOS and PDOS of a-Fe(C)-M.
态密度图中,仅能反映a-Fe(C)-M超晶胞中的原子参与成键情况,至于成键的类型以及成键的强弱还需要通过Mulliken电荷布居和重叠布居进行分析.
表3为a-Fe(C)-M的Mulliken电荷布居.可以看出,Zr,Nb,V固溶于铁素体后,Fe,Zr,Nb,V原子均表现为失电子,而C表现为得电子,说明合金原子中有一部分电子向C原子发生了转移与C原子形成了离子键.表4为a-Fe(C)-M的重叠电荷布居数[28],聚居数为正值表示存在共价键,聚居数为负值表示原子间存在反键,原子间相互排斥,聚居数为0则表示存在离子键,聚居数值越大,则键的作用越强.从表4中a-Fe(C)-Zr的数据可以看出,C—Fe键聚居数为3.54,C—Zr键聚居数为0.11,均为正值,因此C—Fe键、C—Zr键均表现为共价键,但C—Zr共价键作用较弱.结合图2(a)中的分析可以得出,Zr固溶于铁素体后,形成了Fe—C键和Zr—C键,其中Fe—C键为共价键,Zr—C键以离子键为主,同时也形成了较弱的共价键.同样的分析得出,Nb固溶于铁素体后,形成了Fe-C键和Nb—C键,其中Fe—C键为共价键,Nb—C键以离子键为主,同时也形成了较弱的共价键; V固溶于铁素体后,形成了Fe—C键、Fe—V键和C—V键,其中Fe—C键、C—V键均为共价键,Fe—V键为离子键.
从表3中可以看出,Zr,Nb,V固溶于铁素体形成离子键,从电子转移数量上看,V转移的电子数是为0.59,是三种合金原子中最多的,Zr与Nb电子转移数量相差不大.电子转移的数量越多,离子键的相互作用就越强,因此V与铁素体晶胞中原子的离子键作用最强,Zr,Nb与铁素体晶胞中原子的离子键作用相差不大且相对较弱.表4中,重叠聚居数为正值,值越大则共价键作用越强.因此V与铁素体晶胞中原子的共价键作用最强(聚居数为3.81),Nb次之而Zr的共价键作用最弱.

表3 a-Fe(C)-M的Milliken电荷布居Table 3.Milliken charge of a-Fe(C)-M.
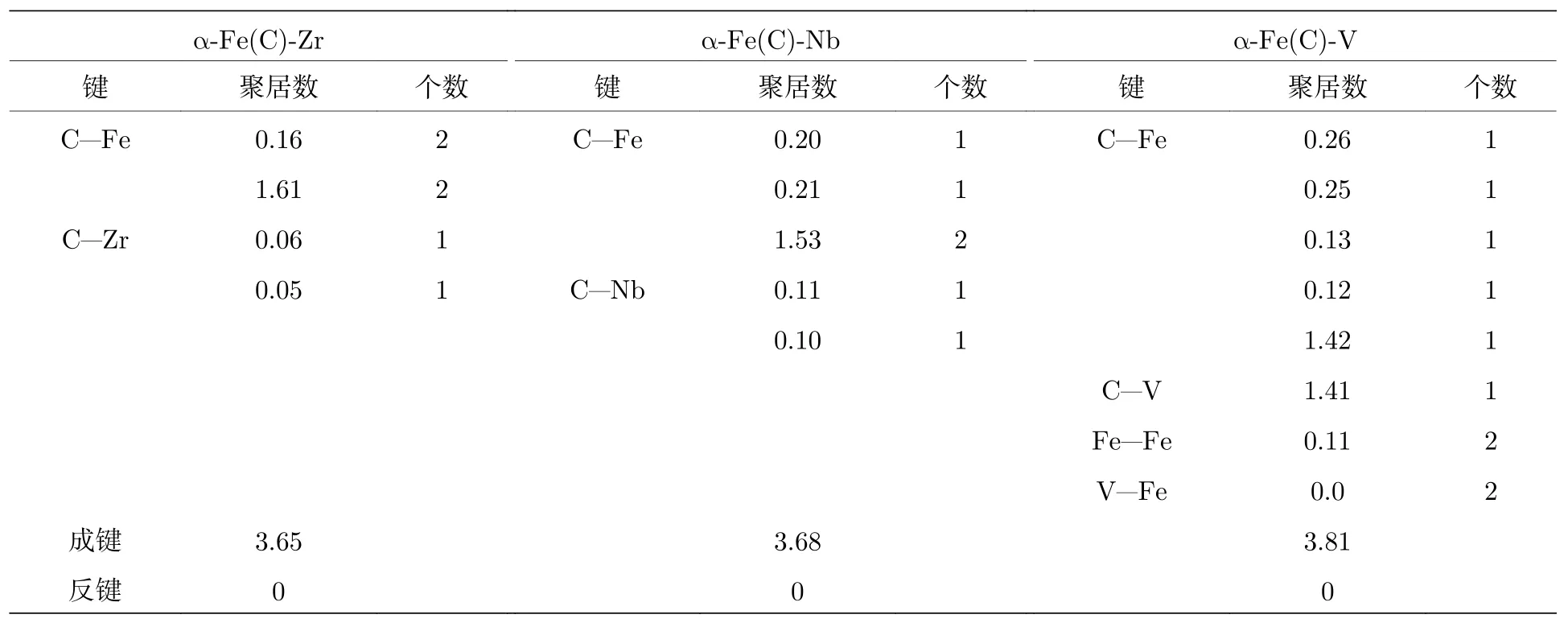
表4 a-Fe(C)-M的重叠电荷布居Table 4.Overlap charge distribution of a-Fe(C)-M.
综合以上分析得出,Zr,Nb,V固溶于铁素体后,Zr,Nb仅与Fe原子形成金属键,而V与铁素体晶胞中的Fe原子形成了金属键及Fe-V离子键,其中离子键的作用强于Zr,Nb原子与铁素体晶胞中的键合作用,使V固溶于铁素体后,晶胞的结合能增加,稳定了铁素体.
3.4 力学性能分析
a-Fe(C)-M晶胞的力学性能(体模量B、剪切模量G、杨氏模量E以及泊松比s)可以根据所得的弹性常数进行计算.a-Fe(C)-M晶胞经结构优化后,空间点群为四方晶系P4/mmm,晶系的弹性常数有六个独立变量,即C11,C12,C13,C33,C44和C66.由这些弹性常数,采用(2)式—(4)式[29]即可获得a-Fe(C)-M晶胞的力学性能.
计算结果如表5所列.一般来说,材料硬度的半经验公式与杨氏模量E和剪切模量G存在一定关系[30],这是因为试验硬度测量值与压痕深度有关,其中剪切模量G对压痕阻力的恢复起主要作用.杨氏模量E又与剪切模量G密切相关,即增加剪切模量G和杨氏模量E可以提高材料的硬度.对于大多数材料而言,提高硬度的同时还要保持一定的延展性.根据Pugh材料脆性/延展性判据[31],材料的体模量与剪切模量之比B/G> 1.75为韧性材料,否则为脆性材料.另外,泊松比也可以反映材料的塑性性能,泊松比s越大材料的塑性就越好.
从表5中可以看出,Zr,Nb固溶后,B,G,E,s均无明显变化,表明Zr,Nb的固溶对合金铁素体的力学性能影响不大.V固溶后,体模量B增加,剪切模量G和杨氏模量E大幅降低,这表明V原子的固溶使a-Fe(C)的硬度降低但抗压强度有所提高,可以认为V的固溶对a-Fe(C)起到强化作用.这是因为体模量B与晶体原子的结合能和键能有关[32].体模量B越大,意味着原子间的结合能越大,键的强度越强,能够起到固溶强化作用.正是由于Zr,Nb,V原子半径不同,导致V原子与晶胞Fe,C原子的原子间距相对较小,这是晶胞结合能增加的主要原因.因此,V固溶后对晶胞主要起到提高韧性的作用.
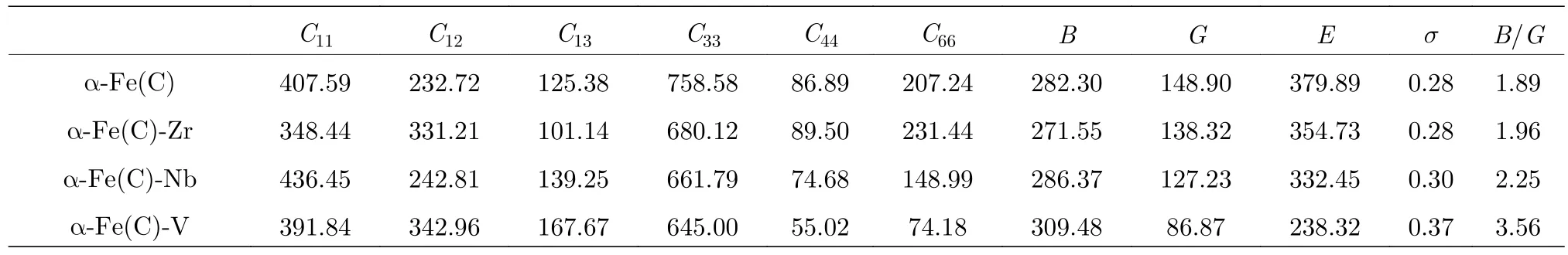
表5 a-Fe(C)-M晶胞的弹性常数及力学性能Table 5.Elastic constants and mechanical properties of a-Fe(C)-M.
4 讨 论
钢的微合金化技术是改善钢铁材料力学性能的重要方法之一.虽然Zr,Nb,V均是强碳化物形成元素,但由于其与C的亲和力不同,因此添加微量Zr,Nb,V对改善钢铁材料力学性能的机理有所不同.第一性原理计算表明,由于Zr,Nb,V原子半径及电子结构的差异,其固溶于a-Fe(C)的占位不同并使晶胞获得了不同的结合能.Zr,Nb元素固溶于a-Fe(C)后降低了晶胞的结合能,因此Zr,Nb更倾向于与C形成ZrC,NbC强化相,一方面增加了基体本身的硬度,另一方面,强化相也能够阻碍奥氏体晶粒的长大,细化晶粒而提高材料的强度及硬度.因此Zr,Nb主要是通过弥散强化的方式改善钢铁材料的力学性能.少量的V能够固溶于a-Fe(C),提高晶胞的结合能,提高铁素体的稳定性.V固溶后,能在一定程度上提高铁素体的韧性,是提高力学性能的主要原因.
5 结 论
论文采用第一性原理的计算方法,计算了Zr,Nb,V在a-Fe(C)中的晶胞总能、结合能,并计算了a-Fe(C)-M晶胞的态密度、电荷布居数以及力学性能,主要得到了以下结论:
1)晶格体积变化率和晶胞总能的计算结果表明,V优先置换a-Fe(C)晶胞中顶角位置的Fe原子,而Zr,Nb优先置换a-Fe(C)晶胞中体心位置的Fe原子;
2)结合能及力学性能的计算结果表明,Zr,Nb降低了铁素体的稳定性,Zr比Nb更难固溶于a-Fe(C).V固溶后增加了晶胞结合能,对晶胞主要起到提高韧性的作用;
3)态密度、电荷布居及重叠电荷布居数的计算结果表明,Zr固溶于铁素体后,形成了Fe—C键和Zr—C键,其中Fe—C键为共价键,Zr—C键以离子键为主,同时也形成了较弱的共价键.Nb固溶后的成键类型与Zr固溶后的原子成键类型相同.V固溶于铁素体后,形成了Fe—V键和C—V键,其中C—V键为共价键,Fe—V键为离子键.其中离子键的作用强于Zr,Nb原子与铁素体晶胞中的键合作用是晶胞结合能增加的主要因素;
4)Zr,Nb主要是通过弥散强化的方式改善钢铁材料的力学性能,V固溶后,能在一定程度上提高铁素体的韧性,是提高力学性能的主要原因.