染色体10q24.31片段重复导致先天性缺指/缺趾畸形的一个家系致病机理分析
2019-08-26张秀泉王建熊符吕伟标周远青杨少民张玉婷田小燕连蔚徐湘民
张秀泉,王建,熊符,吕伟标,周远青,杨少民,张玉婷,田小燕,连蔚,徐湘民
染色体10q24.31片段重复导致先天性缺指/缺趾畸形的一个家系致病机理分析
张秀泉1,2,王建3,熊符3,吕伟标1,周远青1,杨少民4,张玉婷2,田小燕2,连蔚2,徐湘民3
1. 南方医科大学顺德医院检验科,生殖遗传研究室,佛山 528300 2. 南方医科大学顺德医院妇产科,佛山 528300 3. 南方医科大学医学遗传学教研室,广州 510515 4. 南方医科大学顺德医院放射科,佛山 528300
先天性缺指/缺趾畸形表现为手足指/趾骨发育不全等症状,会严重影响患者生活中的精细操作及心理健康。本研究对一个患有先天性缺指/缺趾畸形家系进行了基因变异检测,分析总结了该疾病分型与基因变异之间的关联关系,并探讨了对此类疾病患者开展遗传咨询及基因诊断的策略。首先,采用临床体检及四肢X线检查的方式,对患者表型进行分析。然后,应用D10S1709、D10S192、D10S597、D10S1693和D10S587等5个位点对外周血DNA进行了单倍型分析,并利用Array-CGH检测基因重复片段。最后,通过基于家系调查和基因分析探讨先天性手足裂畸形的致病原因。研究结果显示,先证者为典型的先天性手足裂畸形,表现为双侧食、中指缺失,拇指短,左手无名指畸形与缺失中指的皮肤相连成蹼状;双足正中裂开至足中部,第2和3趾缺失,第4和5趾融合。家系中其他患者表型变异较大。其外周血基因单倍型分析表现为染色体10q24.31-10q24.32区域有一个至少610 kb的重复,Array-CGH分析结果为10q24.31(102 832 650~103 511 083)×3。对先证者及其弟弟和父母进行单倍型分析,确认该家系的致病基因为10q24.31-10q24.32基因重复,单倍型165-251-289-219-102为该病的等位基因。研究结果提示,该家系缺指/缺趾畸形乃由于染色体10q24.31 (102 832 650~103 511 083)×3引起,其单倍型165-251-289-219-102可作为检测10q24.3110q24.32等位基因的疾病标 志物。
先天性手足裂畸形;微阵列比较基因组杂交技术;单体型基因分析;遗传咨询
先天性缺指/缺趾畸形(congenital ectrodactyly)是指出生前肢体有手指和/或脚趾的缺失,临床可表现为手指/脚趾缺失、缺失+并指/趾、手掌/脚掌分裂,也可多种情况并存。其中一种较严重的情况表现为四肢正中轴端骨发育不全而剩余端骨呈不同程度的融合,临床表现为手足正中裂开、指骨/趾骨发育不全及并指/趾畸形,称为手足裂畸形(split hand/foot malformation syndrome, SHFM)[1],该病严重地影响患者的精细活动和外观及心理健康,一些患者可伴有外胚层和颅面发育不良、智力低下和口面部裂等情况[2],我国发病率约为1.64/10 000[3],高于国外的1/25 000~1/8500[4]。其遗传方式多为常染色体显性遗传,也可表现为常染色体隐性遗传和X连锁遗传等方式。目前已经报道了7种先天性手足裂畸形与多个基因变异有关[1]。本文主要报道了一个患SHFM家系的遗传学分析结果、该综合征的异常畸形特点及遗传关联因素的最新进展,探讨了生殖健康的基因诊断方法和策略。
1 对象与方法
1.1 对象及临床资料收集
先证者为男性,28岁,汉族,因家族中有多名手足裂畸形患者,想生育健康孩子,来我院生殖医学中心就诊。其妻子正常。
通过临床问询、体格检查收集先证者及其家系成员的临床资料,家系中共有5代22人,6例患者,男女都有发病,其中男性患者4例、女性患者2例,分别是先证者的弟弟、母亲、大舅、祖父及曾祖母,其中祖父和曾祖母已故(图1),其余家系成员正常。对部分不能来检查的或不在世的亲属进行电话问询或请近亲介绍情况。对先证者的双手手掌、尺桡骨和肘腕关节及双脚、胫腓骨和膝踝关节进行了全面的X线拍片检查,包括平面、斜面及侧面,以了解相关骨骼的发育。对夫妻双方进行了常规染色体 检查分析。同时对先证者及父母、弟弟进行了基因分析,其大舅在加拿大,且后代正常,故未做基因分析。
本研究获南方医科大学顺德医院医学伦理委员会论证批准,患者签署了知情同意书。
1.2 外周血全基因组DNA提取
采集患者及其妻子、父母和弟弟的肘前静脉血各2 mL,EDTA抗凝,外周血白细胞基因组DNA提取采用试剂盒(QIAamp DNA blood Midi Kit,德国Qiagen公司),提取方法按照说明书进行。用Nanodrop 2000型核酸蛋白检测仪测定A260和A280,检测DNA的纯度。
1.3 微阵列比较基因组杂交(array-CGH)技术检测
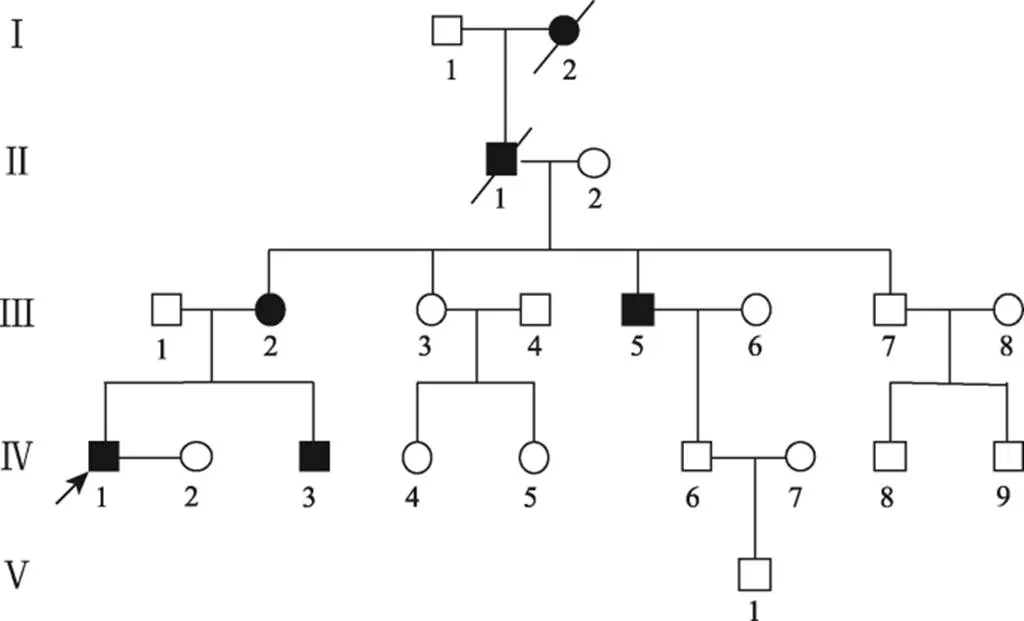
图1 SHFM患者家系图
该家系共调查了5代共有6例患者,男女都有发病,共有男性患者4例、女性患者2例。箭头指向先证者。
患者全基因组DNA进行array-CGH分析应用Affymetrix CytoScan HD Array芯片(国家和公司 名称),该芯片包含有750 000 SNP和1 900 000 CNV标记。操作过程按说明书进行。检测结果用Affymetrix@Chromosome Analysis Suite 2.0 (ChAS 2.0)软件进行分析。
1.4 单倍型基因位点分析
外周血DNA抽提后,应用荧光标记物探针对先证者及其弟弟和父母染色体10q的5个位点进行了单倍型分析,这5个标记物分别是:D10S1709、D10S192、D10S597、D10S1693和D10S587 (表1)。
2 结果与分析
2.1 SHFM患者临床表现
先证者患者临床表现为:双侧食指、中指缺失,双侧拇指短,左手无名指畸形,中部与缺失中指的皮肤相连成蹼状(图2A);双侧大脚趾和脚掌自中部开始分开,第二趾和第三趾均缺失,第四和第五脚趾融合形成了一个畸形的脚趾(图2B)。先证者弟弟出生时左手六指畸形,左手食指末端内弯畸形伴指甲缺失;右脚中趾缺失畸形,大拇趾与第二趾并趾畸形,第四趾肥大,小趾缺趾甲;左脚母趾内弯畸形,余未见异常。先证者母亲双手拇指弯曲畸形,双手食指末端指甲缺失;双脚大拇趾内弯畸形,左脚二、三、四趾短小畸形且无趾甲,右脚中间三指缺失畸形。
2.2 X光检查结果
先证者X光检测结果显示:右手第二掌骨、第一指骨(拇指)、第二指骨(食指)及第三指骨(中指)缺如,第三掌骨轻度发育不良。左手食指、中指中远节指骨缺如,拇指远节指骨、食指近节指骨、环指远节指骨发育不良,中指近节指骨发育不良并与环指近节指骨并指畸形。双足均为第二、三跖骨及第二、三、四趾骨缺如,第四、五跖骨并指畸形。双侧腕/踝关节、尺桡骨及胫腓骨未见异常(图2,A和B)。

表1 荧光探针标记物位点
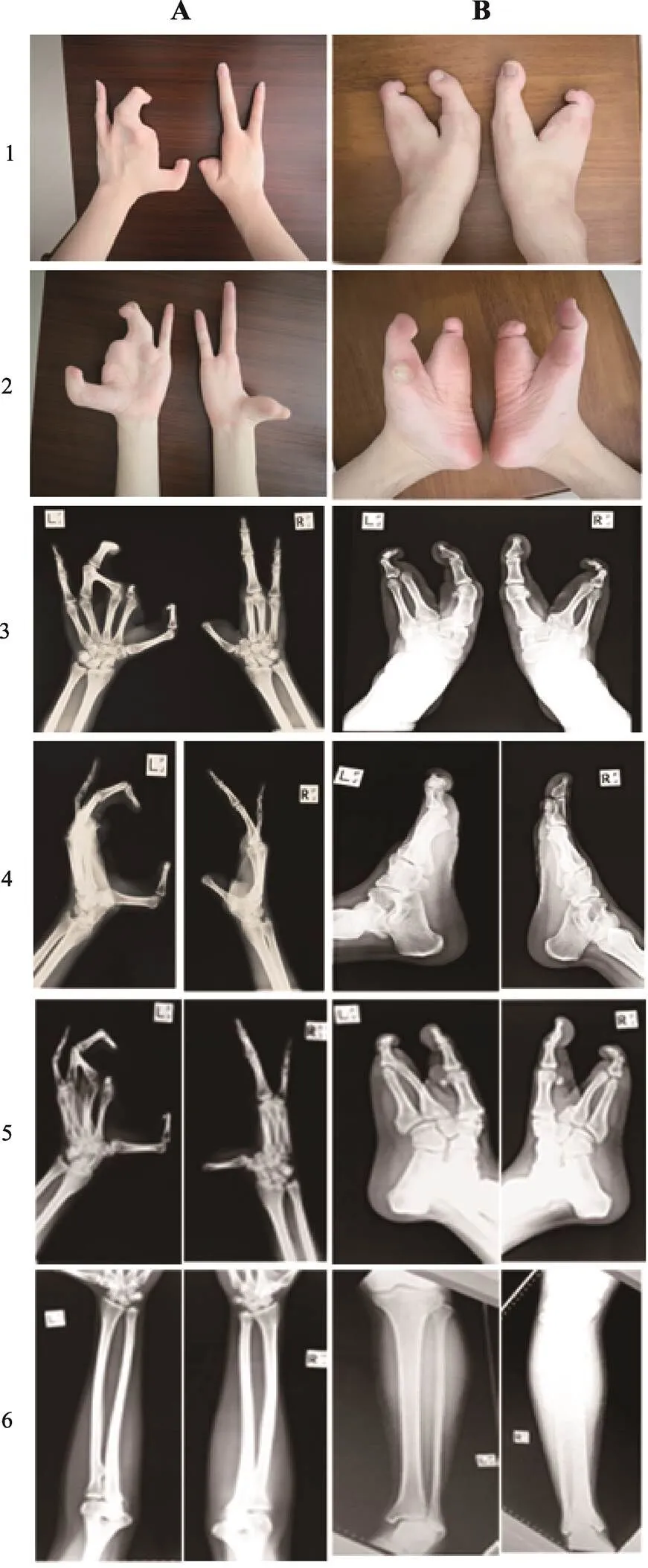
图2 先证者手足照片及X光片
A:患者双手外观表型和X光检测结果;B:患者双脚外观表型和X光检测结果。A1:双手背面,B1:双脚背面;A2:双手掌面,B2:双脚掌面;A3、A4、A5分别为双手X光平面、斜面及侧面,B3、B4、B5分别为双脚X光平面、斜面及侧面;A6为双手尺桡骨及肘腕关节,B6为双脚胫腓骨及膝踝关节。
2.3 Array-CGH基因检测结果
对患者基因组进行array-CGH进行基因比对分析,检测结果发现患者为10号染色体长臂存在一个片段重复(3个拷贝),为10q24.31 (102 832 650~103 511 082)×3 (图3)。
2.4 单倍型基因检测结果
对先证者及其弟弟和父母,本研究采用荧光标记探针对其外周血DNA进行了5个位点(D10S1709、D10S192、D10S597、D10S1693和D10S587)的单倍型分析,检测结果见图4。结果分析表明,母系单体为165-251-289-219-102,父系单体遗传为167-251-291-219-109。结合临床表型家系图和基因分析结果,确认该家系的致病基因为10q24.31- 10q24.32基因重复。因此,单倍型165-251-289-219- 102可作为该病的等位基因诊断标记用于导致该病的10q24.31-10q24.32重复的生殖遗传学诊断。
3 讨论
3.1 SHFM分型及其遗传连锁
SHFM的遗传方式多表现为常染色体显性遗传,也可以表现为常染色体隐性遗传或X连锁遗传。目前为止有7种类型报道,其中1、3、4、5型为常染色体显性遗传[1,3,4],6型为常染色体隐性遗传[5],2型为X连锁遗传[6],SHFLD表现为不完全显性遗 传[7](表2)。
SHFM-Ⅰ型:遗传位点在7q21.2-q21.3,属典型的常染色体显性遗传,可以是7q21区域的缺失、易位或倒置[8]。据文献报道,该区域的和基因缺失或表达不足会导致SHFM。可表现为外显率降低或综合征肢体畸形综合征[9]。约1/3的患者出现感音神经性耳聋,而足外胚层发育不良、唇腭裂的发生率较低[10]。
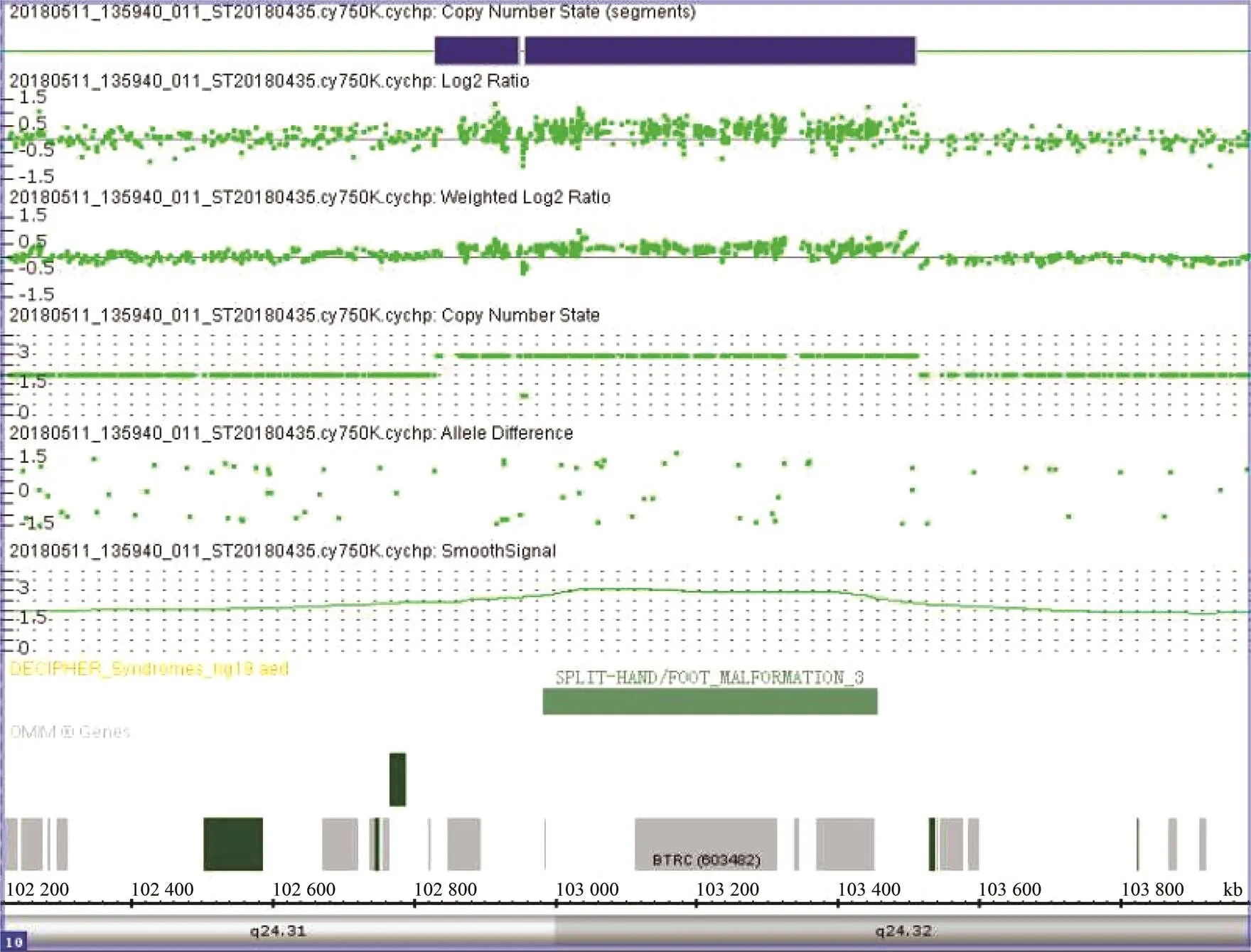
图3 10q24.31染色体片段重复扫描结果
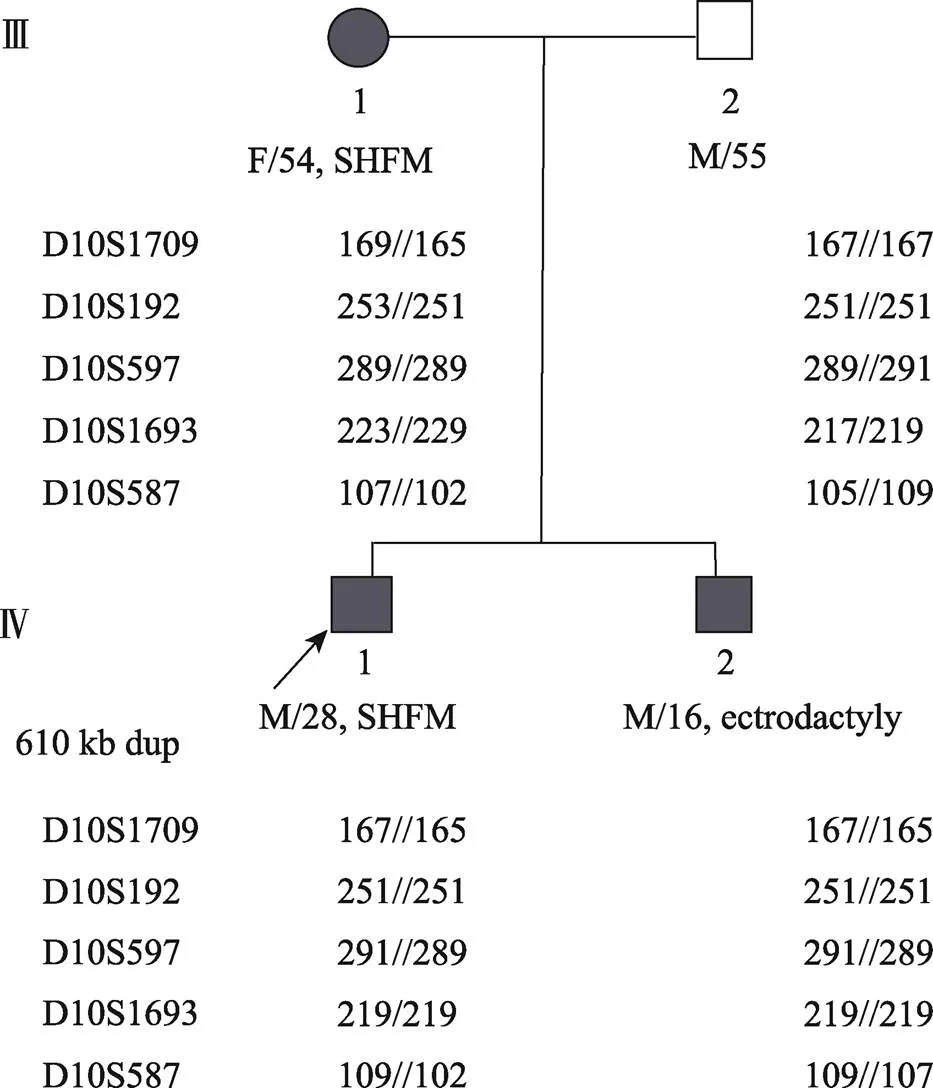
图4 先证者兄弟及其父母5个位点单倍型分析结果
先证者(箭头指向)母系遗传位点为251-102-289-219-165,其弟弟母系遗传位点为251-107-289-219-165,他们的父系遗传位点均为: 251-109-291-219-167。
SHFM-Ⅱ型:遗传位点在Xq26,为X连锁遗传[6]。致病基因与和变异有关,但突变的遗传密码尚未得到证实,精细的基因定位定义了一个5.1 Mb区域,其基因靠近着丝粒,基因靠近端粒。受影响家庭中男性患者多于女性,女性患者可为部分外显。
SHFM-Ⅲ型:遗传位点为染色体10q24微小片段重复,为显性遗传[6]。随后,该区域的几个基因被认为与手足裂畸形的发生有关。利用单核苷酸多态性(SNP)芯片鉴定出10q24.31-q24.32(chr.10:102 962 134~ 103 476 346,hg19)处514 kb的增益,导致了包括和在内的基因重复。除SHFM外,可表现出小颌畸形、听力问题和肾发育不良[1]。
SHFM-Ⅳ型:遗传位点为3q27,为显性遗传,致病基因是突变。基因图谱分析发现,基因突变是SHFM致病基因中唯一的单基因显性遗传位点[11]。在外胚层刺发育中起着关键作用,其异常表达可导致外胚层来源的发育不良[12]。突变可能与10%~16%的SHFM相关,与93%的EEC相关(ectrodactyly ectrodermal dyspasia cleftlip/palate)[2]。
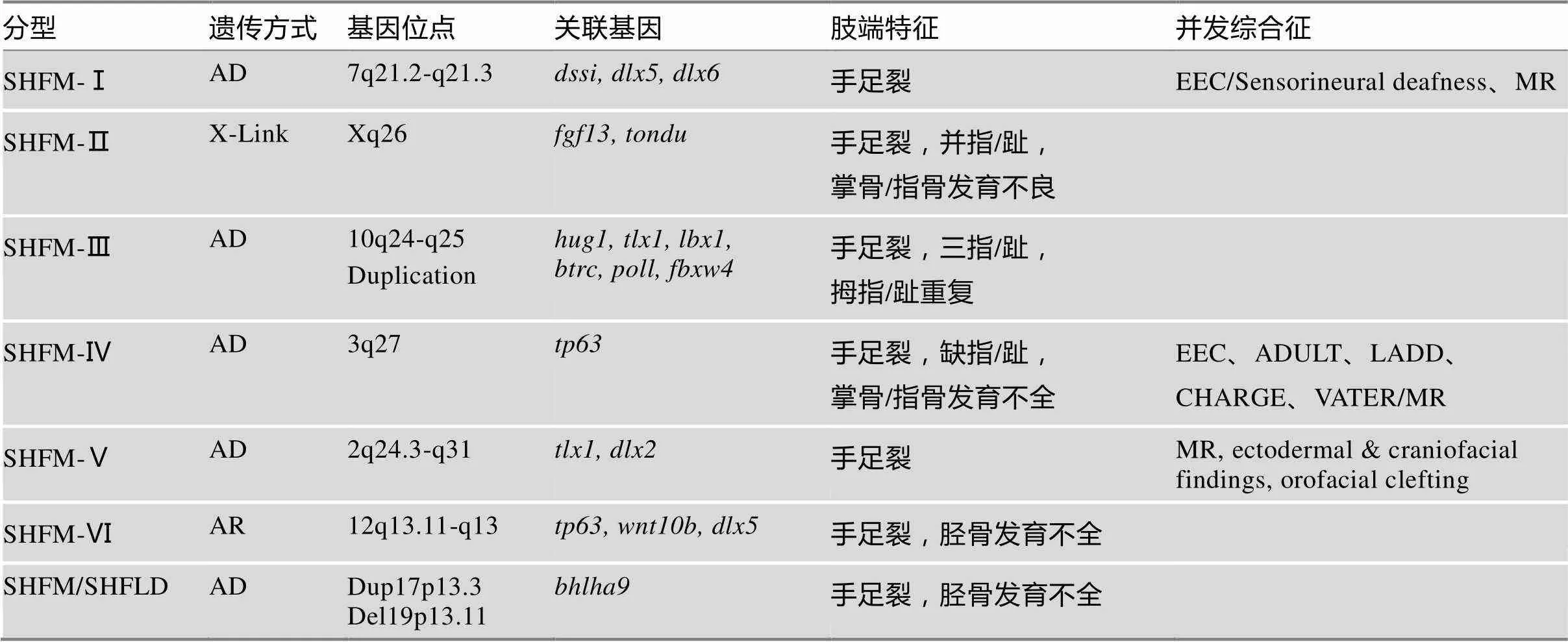
表2 先天性手足裂畸形分型及其遗传连锁
AD:常染色体显性遗传(autosome dominant);AR:常染色体隐性遗传(autosome recessive);SHFM:手足裂畸形(split hand/foot malformation syndrome);SHFLD:手足裂畸形伴长骨发育不全(split hand/foot malformation with long bone deficiency);EEC:缺指畸形伴唇颚裂(ectrodactyly-ectoderma dysplasia-cleft lip/palate);ADULT:肢端皮肤泪牙综合征(acro-dermato-ungual-lacrimal-tooth syndrome);LADD:泪耳综合征(lacrimo-auriculo-dento-digital dyndrome);CHARGE:干眼、心脏畸形,鼻腔狭窄、发育迟缓、泌尿生殖器异常、耳聋综合征(coloboma of the eye, heart defects, atresia of the nasal choanae, retardation of growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness);VATER:脑室异常、肛门闭锁、心血管畸形、食道气管瘘管、肢端缺陷(vertebral anomalies, anal atresia, cardiovascular anomalies, tracheoesophageal fistula, renal and/or radial anomalies, limb defects);MR:智力发育迟缓(mental retardation)。
SHFM-Ⅴ型:遗传位点2q24.3-q31,为显性遗传,关键区域的基因变异包括和基因[13],但该区域的基因突变影响上游距着丝粒5 Mb的基因,与多指/多趾畸形有关[14]。临床表现与其他SHFM类型相比差异很大[15]。
SHFM-Ⅵ型:遗传位点12q13.11-q13,为隐性遗传,3个基因()的7个位点变异与SHFM-Ⅵ型有关[16,17]。纯合子变异(.)是导致SHFM的关键原因,此外,突变亦可导致了相同的综合征[18,19]。
SHFLD (split hand/foot malformation with long bone deficiency)型:手/足裂畸形伴长骨发育不全,其遗传位点在17p13.3[20],为显性遗传,被证实是一个基因重复,但有表型外显减弱的倾向,表现为手足分裂畸形合并胫/腓骨短促[10,21]。动物实验证实了这个基因在胫腓骨发育中的作用。另外,其他作者报道了19p13.11缺失导致SHFM,其基因变异是该疾病的根本原因[22]。
3.2 染色体10q24.3区域基因重复导致SHFM
本研究涉及的SHFM家系,男女均有患病,每代均有患者,系谱分析显示为常染色体显性遗传。通过对家系的临床表现及基因检测结果分析,结果为染色体10q24.31重复,提示为SHFM-Ⅲ型。其家系中患者临床表现类似,个体间有差异,可见其临床表型有异质性差异。基因分析的结果为进行临床遗传咨询和其家系健康生育规划的指导提供了准确的依据。
SHFM-Ⅲ型的遗传致病基因位于染色体10q24- q25,为常染色体显性遗传,研究报道10q24.3区域DNA重复与手足裂畸形的发生有关,表型变异可以较大[23,24]。临床表型可为部分指/趾缺失和指/趾发育不良,以中轴线表现居多。基因重复区域可能包括和基因,以及这些基因的部分重复[1,25]。
先证者的母系遗传单倍体检测结果(165-251- 289-219-102)显示与其弟弟的母系遗传单倍体结果(165-251-289-219-107)有一个基因位点差异。然而,在表型表现上,先证者有典型的SHFM,表现为末端肢体中轴严重畸形,发育不良,第二和第三指/趾发育不良,手足正中分裂。与先证者的表型相比,其弟弟的畸形较小,只有第三右中趾发育不良和第一和第二右趾融合。由此提出一个问题,那就是102位点是不是胚胎发育过程中形成典型SHFM的重要位点?还是环境因素在胎儿早期起着重要作用?从目前的研究中尚还无法得出结论,也许需要进一步利用转基因动物进行研究来回答这一问题。总之,根据临床表型调查和遗传分析,本研究确定10q24.31 (102 832 650~103 511 083)重复是该家族发生SHFM的原因,251-102/107-289-219-165位点是诊断标记位点。
3.3 生殖健康策略探讨
尽管分子生物学遗传诊断技术的开发和推广应用为先天性遗传病患者包括SHFM患者的基因诊断提供了精准诊断的方法,但由于SHFM类型较多,涉及的基因变异较广,对患者的临床咨询和诊断仍然是较大的挑战,特别是对有生育要求的患者家系进行临床遗传咨询及指导,保证下一代的健康生殖生育显得格外重要。首先,要让患者了解这一疾病的自然遗传规律并给他们提供预防和临床处理的有效方法;其次,要给患者家庭提供健康生殖生育的可靠技术保障;最后,要排除可能引起这一疾病的潜在致病遗传因素。
本研究报道的家系现已诊断明确,其SHFM发病是由于染色体10q24.31(102 832 650~103 511 083) ×3引起,其单倍型165-251-289-219-102/107可作为检测10q24.3等位基因的疾病标志物。目前可用的比较可靠的手段帮助SHFM患者或其家庭成员避免遗传该病给子代的方法有种植前遗传学诊断和产前遗传学诊断。
种植前遗传学诊断(preimplantation genetic diagnosis, PGD)对于已知致病基因的SHFM患者家系,在怀孕前进行植入前遗传学诊断,可以大大降低后代的发病风险。单基因疾病的PGD可通过对体外受精(fertilization, IVF)胚胎的遗传学检测,确定胚胎是否带有致病基因,转移未受影响的胚胎,这一技术对于所有已知致病基因的SHFM患者或基因携带者都适应,只要已经鉴定出了特定的家族性突变,就可为其制备针对这一基因的特定检测方法。
PGD的临床过程包括:(1)个案分析,即与准父母交谈并讨论是否需要对夫妻或其他家庭成员进行额外的基因检测;(2)实验室诊断方法建立,即针对患者家庭的特定致病遗传基因制备探针并建立实验室遗传学诊断方法;(3)体外受精和胚胎培养,即通过排卵周期监控和调节并将获得的成熟精卵在体外受精并培养成早期胚胎;(4)胚胎活检,即在胚胎的3~5天利用吹吸的方法获取1个或数个胚胎细胞用作遗传学诊断;(5)遗传学检测,即对获取的胚胎细胞进行已知致病基因的遗传学检测,确定携带和未携带致病基因的胚胎;(6)胚胎移植,即将未带致病基因的胚胎适当适时移植。通过这一系列过程,可以使遗传性患病家庭避免怀上携带致病基因的胚胎,从而获得健康下一代。
产前遗传学诊断(prenatal genetic diagnosis)对于已知致病遗传基因的SHFM患者或携带者,在怀孕早期或中期获取胎儿细胞进行遗传学诊断,可以发现胚胎或胎儿是否携带致病基因,从而帮助孕妇决定是否继续妊娠[26]。在胚胎8~10周或妊娠10~12周,可以通过抽吸绒毛获取遗传学的检测组织;在怀孕14~20周,可以抽取羊水获取脱落的胎儿细胞;在妊娠20~24周,可经皮抽取胎儿脐带血。这3种方法是有创性的,可能产生并发症,其流产或早产的发生率为1%~2%,但作为生殖健康的重要诊断方法,产期遗传学诊断仍然是我国主要的防治遗传性疾病的重要手段。
[1] Gane BD, Natarajan P. Split-hand/feet malformation: a rare syndrome., 2016, 5(1):168–169.
[2] Enriquez A, Krivanek M, Flottmann R, Peters H, Wilson M. Recurrence of split hand/foot malformation, cleft lip/palate, and severe urogenital abnormalities due to germline mosaicism for TP63 mutation., 2016, 170(9):2372–2376.
[3] Dai L, Li YH, Deng Y, Zhu J, Wang YP, Liang J, Zhang YW, Liu ZY. Prevalence of congenital split hand/split foot malformation in chinese population.,2010, 41(2): 320–323.代礼,李艳华,邓莹,朱军,王艳萍,梁娟,张亚玮,刘芝英. 中国人群裂手裂足畸形的流行病学特征.四川大学学报(医学版), 2010, 41(2):320–323.
[4] Bedard T, Lowry RB, Sibbald B, Kiefer GN, Metcalfe A. Congenital limb deficiencies in Alberta-a review of 33 years (1980-2012) from the alberta congenital anomalies surveillance system (ACASS).,2015, 167A(11):2599–2609.
[5] Kantaputra PN, Kapoor S, Verma P, Intachai W, Ketudat Cairns JR. Split hand-foot malformation and a novel WNT10B mutation., 2018, 61(7):372–375.
[6] Faiyaz-Ul-Haque M, Zaidi SH, King LM, Haque S, Patel M, Ahmad M, Siddique T, Ahmad W, Tsui LC, Cohn DH. Fine mapping of the X-linked split-hand/split-foot malformation (SHFM2) locus to a 5.1-Mb region on Xq26.3 and analysis of candidate genes., 2005,67:93–97.
[7] Fusco C, Nittis P, Alfaiz AA, Pellico MT, Augello B, Malerba N, Zelante L, Reymond A, Merla G. A new split hand/foot malformation with long bone deficiency familial case, 2016, 6(2):98–102.
[8] Kantaputra PN, Carlson BM. Genetic regulatory pathways of split-hand/foot malformation, 2019, 95(1):132–139.
[9] van Silfhout AT, van den Akker PC, Dijkhuizen T, Verheij JB, Olderode-Berends MJ, Kok K, Sikkema-Raddatz B, van Ravenswaaij-Arts CM. Split hand/foot malformation due to chromosome 7q aberrations(SHFM1): additional support for functional haploinsufficiency as the causative mechanism.,2009, 17(11):1432–1438.
[10] Haberlandt E, Löffler J, Hirst-Stadlmann A, Stöckl B, Judmaier W, Fischer H, Heinz-Erian P, Müller T, Utermann G, Smith RJ, Janecke AR. Split hand/split foot malformation associated with sensorineural deafness, inner and middle ear malformation, hypodontia, congenital vertical talus, and deletion of eight microsatellite markers in 7q21.1-q21.3.,2001, 38(6):405–409.
[11] Dai L, Li NN, Deng Y, Mao M, Wang H, Zhu J. Genotype-phenotype analysis of a Chinese family with split hand/split foot and syndactyly.,2011, 28(4):379–382.代礼, 李娜娜, 邓莹, 毛萌, 王和, 朱军. 一个中国人裂手足伴并指趾畸形家系的表型和基因型分析. 中华医学遗传学杂志, 2011, 28(4): 379–382.
[12] Sharma D, Kumar C, Bhalerao S, Pandita A, Shastri S, Sharma P. Ectrodactyly, ectodermal dysplasia, cleft lip, and palate (EEC syndrome) with tetralogy of fallot: a very rare combination., 2015, 3:51.
[13] Filho AB, Souza J, Faucz FR, Sotomaior VS, Dupont B, Bartel F, Rodriguez R, Schwartz CE, Skinner C, Alliman S, Raskin S. Somatic/gonadal mosaicism in a syndromic form of ectrodactyly, including eye abnormalities, documented through array-based comparative genomic hybridization.,2011, 155A(5):1152–1156.
[14] Goodman FR, Majewski F, Collins AL, Scambler PJ.A117-kbmicrodeletionremovingHOXD9-HOXD13andEVX2causessynpolydactyly., 2002, 70(2):547–555.
[15] Theisen A, Rosenfeld JA, Shane K, McBride KL, Atkin JF, Gaba C, Hoo J, Kurczynski TW, Schnur RE, Coffey LB, Zackai EH, Schimmenti L, Friedman N, Zabukovec M, Ball S, Pagon R, Lucas A, Brasington CK, Spence JE, Sparks S, Banks V, Smith W, Friedberg T, Wyatt PR, Aust M, Tervo R, Crowley A, Skidmore D, Lamb AN, Ravnan B, Sahoo T, Schultz R, Torchia BS, Sgro M, Chitayat D, Shaffer LG. Refinement of the region for split hand/foot malformation 5 on 2q31.1., 2010, 1(5):262–271.
[16] Ullah A, Gul A, Umair M, Irfanullah, Ahmad F, Aziz A, Wali A, Ahmad W. Homozygous sequence variants in the WNT10B gene underlie split hand/foot malformation., 2018, 41(1):1–8.
[17] Aziz A, Irfanullah, Khan S, Zimri FK, Muhammad N, Rashid S, Ahmad W. Novel homozygous mutations in the WNT10B gene underlying autosomal recessive split hand/foot malformation in three consanguineous families., 2013, 534(2):265–271.
[18] Ugur SA, Tolun A. Homozygous WNT10b mutation and complex inheritance in Split-Hand/Foot malformation.,2008, 17(17):2644–2653.
[19] Khan S, Basit S, Zimri FK, Ali N, Ali G, Ansar M, Ahmad W. A novel homozygous missense mutation in WNT10B in familial split-hand/foot malformation.,2012, 82(1):48–55.
[20] Klopocki E, Lohan S, Doelken SC, Stricker S, Ockeloen CW, Soares Thiele de Aguiar R, Lezirovitz K, Mingroni Netto RC, Jamsheer A, Shah H, Kurth I, Habenicht R, Warman M, Devriendt K, Kordass U, Hempel M, Rajab A, Mäkitie O, Naveed M, Radhakrishna U, Antonarakis SE, Horn D, Mundlos S. Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion.,2012, 49(2):119–125.
[21] Al Kaissi A, Ganger R, Rötzer KM, Klaushofer K, Grill F. A child with split-hand/foot associated with tibial hemimelia (SHFLD syndrome) and thrombocytopenia maps to chromosome region 17p13.3., 2014, 164A(9):2338–2343.
[22] Umair M, Ullah A, Abbas S, Ahmad F, Basit S, Ahmad W. First direct evidence of involvement of a homozygous loss-of-function variant in the EPS15L1 gene underlying split-hand/split-foot malformation., 2018, 93(3):699–702.
[23] Dai L, Deng Y, Li N, Xie L, Mao M, Zhu J. Discontinuous microduplications at chromosome 10q24.31 identified in a Chinese family with split hand and foot malformation., 2013, 14:45.
[24] Yang W, Hu ZJ, Yu XF, Li QH, Zhang AJ, Deng X, Zhang AY, Gao CS, Liu Y, Ao Y, Lo WHY, Zhang X. A DNA duplication at chromosome 10q24.3 is associated with split-hand-foot malformation in a Chinese family., 2006, 86(10): 652–658.杨威,胡周军,余晓芬,李气环,张爱菊,邓曦,张爱英,高春生,刘扬,敖杨,罗会元,张学. 中国人手足裂畸形患者中染色体10q24.3区域DNA重复突变的鉴定.中华医学杂志, 2006, 86(10):652–658.
[25] Xiang R, Du R, Guo S, Jin JY, Fan LL, Tang JY, Zhou ZB. Microduplications of 10q24 detected in two Chinese patients with split-hand/foot malformation Type 3.,2017, 47(6):754–757.
[26] Wu LQ, Pan Q, Long ZG, Zhu JZ, Dai HP, Zheng D, Xia K, Huang XQ, Xia JH. Genetic diagnosis and prenatal genetic diagnosis of fragile X syndrome., 2003, 25(2): 123–128.邬玲仟, 潘乾, 龙志高, 朱俊真, 戴和平, 郑多, 夏昆, 黄幸青, 夏家辉. 脆性X综合征的基因诊断与产前诊断. 遗传, 2003, 25(2): 123–128.
Congenital ectrodactyly caused by chromosome 10q24.31 duplication and its pathogenetic analysis
Xiuquan Zhang1,2, Jian Wang3, Fu Xiong3, Weibiao Lv1, Yuanqing Zhou1, Shaomin Yang4, Yuting Zhang2, Xiaoyan Tian2, Wei Lian2, Xiangmin Xu3
In order to investigate the genetic variations and the clinical manifestations of a range of congenital ectrodactyly family and to summarize the split hand/foot malformation (SHFM) types and their related pathogenic genes, we conducted phenotypic analyses of patient’s limbs by physical and X-ray examination. The haplotypes were analyzed by using the extracted genes from peripheral blood on D10S1709, D10S192, D10S597, D10S1693 and D10S587 loci, and the mutation duplication loci were confirmed by Array-CGH detection. The pathogenic factors and inheritance pattern of SHFM were analyzed based on family investigation and gene analysis. Results demonstrate the proband’s phenotype is typically of a congenital SHFM which is manifested by missing bilateral index and middle fingers, short bilateral thumbs, deformed left ring finger with webbing of the skin missing at the middle finger; bilateral big toe with the second and the third toe missing, fourth and fifth toe fusion leading to a deformed toe separated from the first toe by the middle of the foot. The haplotype analyses show that there is a repeat of at least 610 kb in chromosome 10q24.31-10q24.32 region. Array-CGH analysis shows 10q24.31 (102 832 650-103 511 083) ×3. Our results demonstrate that the pathogenic gene variation of ectrodactyly in this family is due to duplication of 10q24.31 (102 832 650~103 511 083). The haplotype 165-251-289-219-102 can be used as a disease marker for detecting 10q24.31~10q24.32 allele for SHFM.
congenital ectrodactyly; array-CGH; haplotype analysis; genetic counseling
2019-05-07;
2019-07-18
南方医科大学顺德医院临床研究基金项目(编号:CRSP2019009)资助[Supported by Clinical Research Fund, Shunde Hospital of Southern Medical University (No. CRSP2019009)]
张秀泉,博士,教授,研究方向: 妇产科生殖遗传。E-mail: zhangxiuquan@yahoo.com
10.16288/j.yczz.19-125
2019/7/9 18:19:11
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190709.1818.003.html
(责任编委: 夏昆)
