养殖大鲵肠道细菌多样性
2019-08-13兰阿峰郭素芬彭浩
兰阿峰 郭素芬 彭浩

摘要:纯培养分离大鲵肠道细菌,选用细菌通用引物进行16S rRNA扩增,测序后提交序列并分析;免培养方法采用试剂盒提取肠道内容物总DNA,选用细菌通用引物对总DNA进行16S rRNA扩增,构建克隆文库,对阳性克隆进行PCR-RFLP分析,用Hae Ⅲ内切酶进行片段插入性验证并进行测序,构建系统发育树。纯培养获得103个菌株,分属于16个不同的可操作分类单元(operational taxonomic units,OTUs),系统发育分析表明这些克隆序列分别属于变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)和放线菌门(Actinobacteria)。其中,变形菌门(占克隆总数 87.63%)为最优势类群。免培养结果将随机挑取的105个阳性克隆归为15个OTUs,系统发育分析表明这些克隆序列分别属于变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、放线菌门(Actinobacteria)和拟杆菌门(Bacteroidetes)4个门。其中,变形菌门(占克隆总数的71.43%)为最优势类群。养殖大鲵肠道细菌多样性丰富,具有进一步开发研究的价值。
关键词:养殖大鲵;肠道细菌;纯培养;免培养;多样性
中图分类号: S966.6 文献标志码: A 文章编号:1002-1302(2019)01-0146-06
中国大鲵(Andrias davidianus)别称“娃娃鱼”,属于两栖纲有尾目隐腮鲵科,是现存个体最大的两栖动物,1998年被列为国家二级保护动物[1]。我国大鲵主要分布在陕西、江西、湖北、四川等17个省(区)深山峡谷的溪流当中[1-3]。中国大鲵是肉食性捕食动物,野生大鲵捕食的动物有螃蟹、昆虫、青蛙、鱼等[4-5]。由于大鲵具有极高的经济价值和药用价值,其人工养殖逐渐得到了重视,目前在陕西汉中、四川巴中等地区出现了较大规模的养殖。
动物肠道寄生着大量的微生物,这些微生物同时也是动物体的组成部分,肠道微生物与寄主消化吸收及健康状况密切相关[6-8]。肠道是一个相对密闭的系统,其内部生长的微生物以厌氧或兼性厌氧的微生物为主,采用传统的培养方法分离依然是肠道微生物研究的主要方法之一,有报道称传统方法分离到的微生物物种只有微生物总量的1%~5%,而其余95%~99%的微生物种群还未被分离和识别[9]。免培养方法是一种基于分子生物学的方法,与传统的分离鉴定方法完全不同,可以弥补传统培养方法的缺点[10]。该方法利用分子生物学技术判断微生物的种类及各个物种之间的亲缘关系,其缺点是无法获取微生物[11-12]。人工养殖由于其养殖密度高致使大鲵容易出现传染性病害,部分传染病与肠道微生物密切相关,因此研究养殖大鲵肠道微生物多样性对人工饲养大鲵保护工作具有理论价值。本研究利用纯培养及免培养2种方法对养殖大鲵的肠道微生物多样性进行研究,以期为大鲵保护工作提供理论依据。
1 材料与方法
1.1 材料
1.1.1 供试材料 养殖大鲵采自陕西理工大学大鲵研究所大鲵养殖室,样本共10条,平均体质量6.76 kg,解剖后取完整消化道放入4 ℃的冰箱中保存,2周内处理完。
1.1.2 主要仪器 超净工作台(江苏苏州净化设备有限公司),高压灭菌器(美国Stik公司),凝胶扫描成像系统(美国Bio-Rad公司),PCR扩增仪(英国Techen公司),高速冷冻离心机(德国Eppendorf公司),移液器(Eppendorf公司),恒温水浴锅(上海精宏实验设备有限公司),超低温冰箱(日本Panasonic公司),核酸测定仪(Eppendorf公司),超纯水净化系统(上海优普实业有限公司)。
1.1.3 主要试剂 Fast DNA Spin Kit For Feces(美国Mo Bio公司),2×Taq PCR Kit(Takara公司),2×HieffTM PCR Master Mix(Takara公司),高效感受態DH5α(Escherichia coli competent cells,DH5α)(上海Sangon公司),pMDTM19-T Vocter Cloning Kit(Takara公司),Spin柱式DNA回收试剂盒(Biofulx公司),PCR引物(上海Sangon公司),Hae Ⅲ(Takara公司),Ampicillin(上海Sangon公司),Trytone(Oxoid公司),Yeast Extract(Oxoid公司)。
1.2 试验方法
1.2.1 大鲵肠道细菌纯培养 纯培养肠道细菌分离,按照文献[13]报道方法进行。将大鲵肠道内容物稀释涂布于牛肉膏蛋白胨平板,37 ℃培养,挑取单菌落进行纯化并保藏菌种于4 ℃冰箱中。
1.2.2 对大鲵肠道纯培养细菌进行分子鉴定 纯培养肠道细菌DNA扩增,参照文献[14]报道方法进行。进行菌液16S rRNA扩增,选用引物799F:5′-AACAGGATTAGATACCCT G-3′/1492R:5′-GGTTACCTTGTTACGACTT-3′进行扩增,长度750 bp左右。反应体系(50 μL):2×Taq Master Mix 25 μL,上下游引物各2 μL,模板DNA 2 μL,ddH2O 19 μL。反应条件:94 ℃ 5 min;94 ℃ 1 min;55 ℃ 0.5 min;72 ℃ 1 min,33 个循环;72 ℃ 10 min。PCR产物用的琼脂糖电泳检测后送上海生工生物工程股份有限公司测序,结果整理后提交NCBI申请大鲵肠道细菌菌株16S rRNA基因序列登录号。
1.2.3 大鲵肠道内容物中微生物总DNA提取及16S rRNA扩增 解剖大鲵后将其肠道内容物收集到灭菌的1.5 mL EP管中。总DNA的提取使用美国Mo Bio公司生产的粪便基因组提取试剂盒。按照试剂盒说明提取DNA后将其干燥,加入20 μL TE缓冲液溶解DNA,-20 ℃保存备用。总DNA特异性扩增,参照纯培养肠道细菌扩增方法进行。
1.2.4 克隆文库的构建及阳性克隆的筛选 用Sanprep柱式DNA胶回收试剂盒将扩增后750 bp左右的目的条带切胶纯化回收。纯化产物与pMDTM19-T Vocter连接,连接产物转化到大肠杆菌DH5α感受态细胞。阳性克隆子的初步筛选参照文献[15],将转化后的细胞涂布含氨苄青霉素(100 mg/L)、X-gal、IPTG的LB琼脂平板37 ℃避光培养 14~16 h进行蓝白斑筛选。对阳性PCR产物选用限制性核酸内切酶Hae Ⅲ对目标插入片段多态性分析(PCR-RFLP)后进行测序。将Hae Ⅲ酶切带谱分析不同,且测序结果也不同的克隆归为1个OTU。克隆文库覆盖率统计分析应用公式:C=[1-(n/N)]×100%[16],这里n代表在16S rRNA基因克隆文库仅出现1次的OTU的数量,N代表16S rRNA基因克隆文库的总数。
1.2.5 免培养获取细菌及克隆文库发育分析 免培养获取的测序结果及克隆文库获取的细菌测序结果用Vector Screen去除载体序列后提交到EzTaxon-eserver(http://eztaxon-e.ezbiocloud.net)进行BLAST检索,下载同源性较高的模式菌株的数据,生成Fasta格式文件。用ClustalX软件对所得序列进行人工校正及比对分析。利用Mega 5.02,按照Neighbor-Joining法聚类,选择1 000个重复做Bootstrap值分析,构建系统发育树。所得的16S rRNA基因序列已提交GenBank数据库,申请16S rRNA基因序列登录号。
2 结果与分析
2.1 大鲵肠道细菌纯培养
2.1.1 大鲵肠道细菌纯培养分离 经牛肉膏蛋白胨培养基,获取103株纯细菌。对获取细菌菌株进行保藏,待分析鉴定。
2.1.2 大鯢肠道纯培养细菌分子生物学的鉴定 应用细菌16S rRNA通用引物799F和1492R对分离得到的细菌进行菌液PCR,得到约750 bp清晰的片段(图1)。可见PCR产物条带清晰,产物量大,可用于测序反应。将750 bp的清晰条带所对应的的PCR产物送上海生工生物工程股份有限公司测序,成功测通103个序列。序列整理后提交GenBank数据库中NCBI并获得养殖大鲵纯培养肠道细菌保守区基因登录号。获取登陆号为KU872287-KU872389。
2.2 大鲵肠道免纯培养及鉴定
2.2.1 肠道内容物总DNA提取及16S rRNA扩增 参照粪便总DNA提取试剂盒的步骤,提取野生大鲵肠道内容物总DNA。0.9%琼脂糖凝胶电泳结果见图2。提取大鲵肠道微生物的总DNA较完整,条带较为清晰,可用于下游试验。应用细菌16S rRNA基因通用引物进行特异性扩增,得到约 750 bp 的目标片段见图3。另外还可看到位于100~200 bp之间较清楚的条带,推测可能为PCR扩增时,退火温度不合适导致大量引物二聚体产生的基因片段。切胶纯化回收位于750 bp的目标条带可避免其影响。
2.2.2 克隆文库的构建及阳性克隆的筛选 将上述750 bp处PCR产物切胶回收后链接pMD-19T载体,并转化至大肠杆菌DH5α感受态细胞内。构建基因克隆文库后获取191个阳性克隆,以质粒通用引物进行菌落PCR扩增,扩增产物用Hae Ⅲ进行插入目的片段的RFLP分析,初步获取酶切带谱不同的克隆后选择105个样品送上海生工生物工程股份有限公司测序。测序结果去除嵌合序列,并排除重复序列后共得到15个OTUs(代表100个有效序列)。根据公式,计算得克隆文库的覆盖率C为 85.00%,可以代表大鲵肠道内生细菌的多样性。
2.3 细菌16S rRNA基因系统发育分析
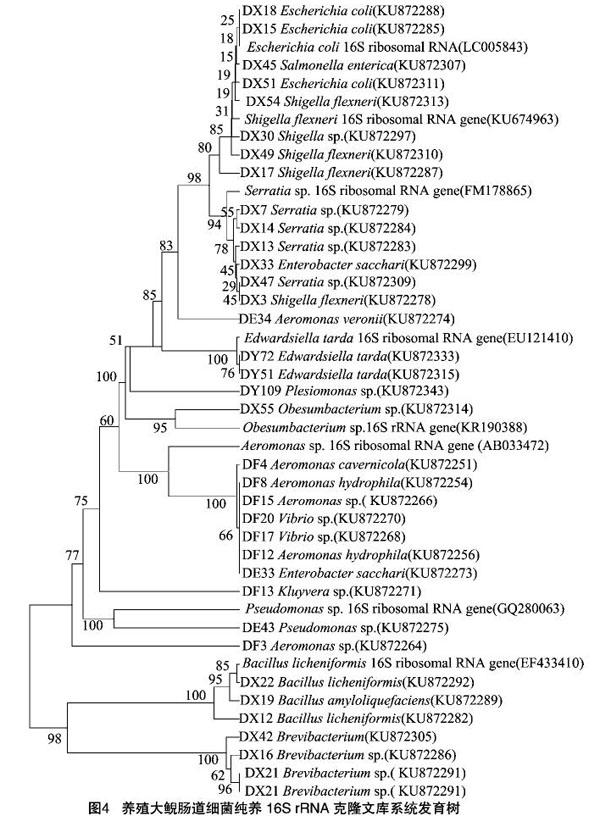
2.3.1 大鲵肠道纯培养细菌16S rRNA基因系统发育分析 纯培养获得103个菌株,成功测通9个序列。整理后提交NCBI数据库,去除嵌合体后得到93条有效序列,登录号为KU872251-KU872389,进行BLAST检索对比,所得到细菌16S rRNA序列如下:变形菌门(Proteobacteria)85株,占总细菌分离数87.63%,其中爱德华氏菌属(Edwardsiella sp.)27株,占总数的27.81%,志贺氏菌属(Shigella sp.)11株占总数的11.34%,沙雷氏菌属(Serratia sp.)5株,占总数的 5.15%,其中埃希氏菌属(Escherichia sp.)8株,占总数的8.25%,沙门氏菌属(Salmonella sp.)2株,占总数的2.06%,肥杆菌属(Obesumbacterium sp.)1株,占总数的1.03%,邻单胞菌属(Plesiomonas sp.)2株,占总数的2.06%,耶尔辛氏菌属(Yersinia sp.)1株,占总数的1.03%,肠杆菌属(Enterobacter sp.)1株,占总数的1.03%,孤菌属(Vibrio sp.)4株,占总数的4.12%,克吕沃氏菌属(Kluyvera sp.)1株,占总数的1.03%,克雷伯氏菌属(Klebsiella sp.)3株,占总数的3.09%,气单胞菌属(Aeromonas sp.)17株,占总数的17.53%,假单胞菌(Pseudomona sp.)2株,占总数的2.06%;厚壁菌门(Firmicutes)占总分离数5.15%,芽孢杆菌属(Bacilli sp.)5株,占总数的 5.15%;放线菌门(Actinobacteria)7株,占总数的7.22%,其中短杆菌属(Brevibacterium sp.)7株,占总数的7.22%。
依据16S rRNA构建的系统发育树(图4),变形菌门(Proteobacteria)作为优势种群,其序列集中在1个大的分支上,而这一分支又分为多个小分支。志贺氏菌属(Shigella sp.)、埃希氏菌属(Escherichia sp.)、沙门氏菌属(Salmonella sp.)在系统发育树上聚为一族。沙雷氏菌属(Serratia sp.)及肠杆菌属(Enterobacter sp.)相似性较高,在系统发育树上聚为一族。爱德华氏菌属(Edwardsiella sp.)与肥杆菌属(Obesumbacterium sp.)相似性较高,在系统发育树上聚为一族。孤菌属(Vibrio sp.)与气单胞菌属(Aeromonas sp.)中的大部分相似性较高,聚为一族。厚壁菌门(Firmicutes)中的短杆菌属(Brevibacterium sp.)占据了进化树的一个独立分支。放线菌门(Actinobacteria)中的短杆菌属(Brevibacterium sp.)占据了进化树的一个独立分支。
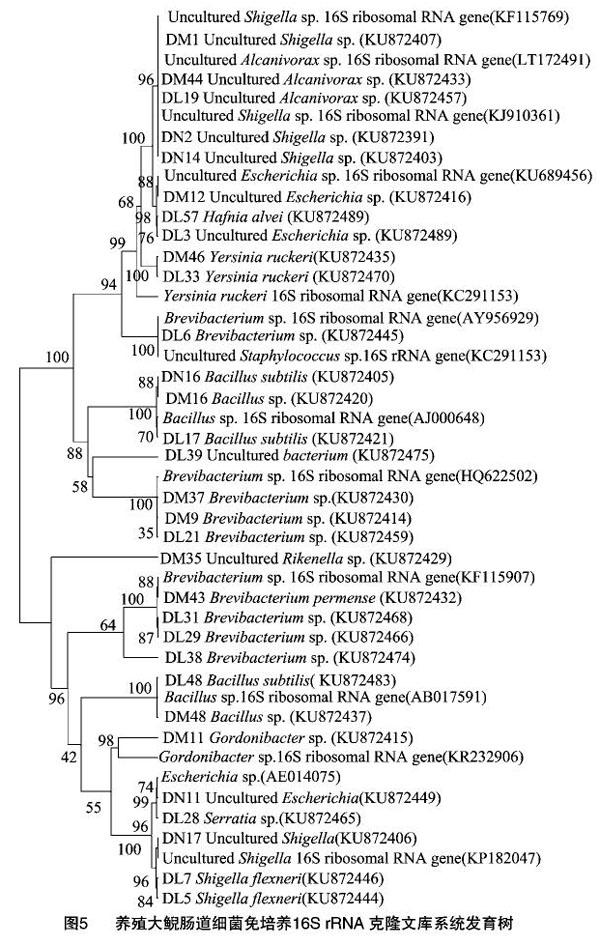
2.3.2 大鲵肠道免培养细菌16S rRNA基因系统发育分析 将测序的105条有效序列,整理后提交GenBank数据库中,取除嵌合体序列获得100个养殖大鲵免培养肠道细菌登录号:KU872390-KU872489。由表2可知,进行BLAST检索后,阳性克隆单元序列(operational taxonomic units,OTU)如下:变形菌门(Proteobacteria)75株,占总克隆数的71.43%,其中埃希氏菌属(Escherichia sp.)29单元,占总数的27.62%,志贺氏菌属(Shigella sp.)30单元,占总数的28.57%,沙雷氏菌属(Serratia)3单元,占总数的2.86%,耶尔辛氏菌属(Yersinia sp.)2单元,占总数的1.90%,食碱菌属(Alcanivorax sp.)7单元,占总数的6.67%,沙门氏菌属(Salmonella sp.)1单元,占总数的0.10%,哈夫尼菌属(Hafnia sp.)1单元,占总数的 0.10%,希万氏菌属(Shewanella sp.)1单元,占总数的 0.10%,布丘氏菌属(Buttiauxella sp.)1单元,占总数的 0.10%;放线菌门(Actinobacteria)21株,占总克隆数的 20.01%,其中短杆菌属(Brevibacterium sp.)19单元,占总数的18.10%,戈登氏杆菌属(Gordonibacter sp.)1单元,占总数的0.10%,红蝽杆菌属(Coriobacterium sp.)1单元,占总数的 0.10%;厚壁菌门(Firmicutes)8株,占總克隆数的7.62%,其中芽孢杆菌属(Bacillus sp.)7单元,占总数的6.67%,葡萄球菌属(Staphylococcus sp.)1单元,占总数的0.10%;拟杆菌门(Bacteroidetes)占总克隆数的0.95%,其中理研菌属(Rikenella sp.)1单元,占总数的0.10%。
依据16S rRNA构建系统发育树(图5),可见进化树明显分为4个大的分支,第1个分支中主要包含了变形菌门(Proteobacteria)大部分种属;第2个分支主要包含了放线菌门(Actinobacteria)的大部分种属;第3个分支只有1个属,是拟杆菌门(Bacteroidetes)的理研菌属(Rikenella sp.);第4个分支主要包含了厚壁菌门(Firmicutes)的大部分种属。
3 讨论与结论
本研究采用纯培养及免培养方法对养殖大鲵肠道细菌多样性进行研究,传统的纯培养方法获取103株细菌,分子生物学方法鉴定为3门16个分类单元。免培养方法获取105条序列,经NCBI上BLAST比对后,鉴定为4门15个分类单元。
笔者所在实验室前期应用免培养技术对野生大鲵肠道细菌多样性进行研究,野生大鲵肠道细菌免培养获得分4个门:变形菌门(Proteobacteria)占总克隆数的92.08%,梭菌门(Clostridia)、衣原体门(Chlamydiae)、芽孢菌门各占总克隆数的1.98%, 其中变形菌门为最优势类群,另外还存在1.98%
细菌未确定到属[15-16]。本研究中养殖大鲵肠道纯培养细菌共分离的103株细菌可分为3个门16个属,在门的分类上优势菌为变形菌门(Proteobacteria),占87.63%,其次为厚壁菌门(Firmicutes)和放线菌门(Actinobacteria);免培养获取105条细菌克隆序列可分为4个门15个属,在门的分类上,优势菌株为变形菌门(Proteobacteria),占总克隆数的71.43%。可见野生大鲵与养殖大鲵肠道细菌在门一级分类中最优势类群均为变形菌门。在属这一级分类上野生大鲵最优势细菌为气单胞菌属(Aeromonas sp.)占总细菌数 66.34%;养殖大鲵免培养研究发现埃希氏菌属(Escherichia sp.)29单元占总数的27.62%,志贺氏菌属(Shigella sp.)30单元占总数的28.57%,这2个属数量基本相当,同为最优势细菌类群;纯培养养殖大鲵肠道细菌最优势菌群为爱德华氏菌属(Edwardsiella sp.)占总数的27.81%。可见在属一级分类上野生与养殖大鲵肠道细菌最优势菌群差异较大,这可能与大鲵的生活环境、摄食的物种种类等有一定的关系。野生大鲵生活在环境温度较低的山涧溪流中,以小型水生动物及昆虫等为食,食性相对比较复杂,而本试验所用养殖大鲵以小鲫鱼为食,食性相对单一,环境也单一。
大鲵为虽然是两栖类动物,但其一般都生活在水里,很少出水活动,所以大鲵生活环境与水生动物更相近。经比较中华绒螯蟹(Brachyura)肠道细菌研究[17],东方鲀(tetraodon fluviatilis)肠道细菌[18],南美白对虾(Penaeus vannamei Boone)肠道细菌[19]与本研究大鲵(Andrias davidianus)肠道细菌多样性的差异,可知它们的肠道细菌优势种群都是变形菌门(Proteobacteria);而陆生环境中的扬州鹅[20]、牛[21]、猪[22]等动物都是厚壁菌门(Firmicutes)为优势群落。可见环境对肠道细菌多样性的影响是明显的。
养殖大鲵肠道细菌多样性丰富,基于纯培养技术可将养殖大鲵肠道细菌归为3门16属,免培养技术可将养殖大鲵肠道细菌归为4门15属。养殖大鲵肠道细菌多样性研究纯培养技术与免培养技术的结果表明在门的差异性较小,在属的差异性较大。就纯培养技术比较养殖与野生大鲵肠道细菌在属的分类上差异性很大。就免培养技术比较养殖与野生大鲵肠道细菌差异性在门与属的分类上都很大。
参考文献:
[1]杨丽萍,蒙子宁,刘晓春,等. 中国大鲵5个野生种群的AFLP分析[J]. 中山大学学报(自然科学版),2011,50(2):99-104.
[2]章克家,王小明,吴 巍,等. 大鲵保护生物学及其研究进展[J]. 生物多样性,2002,10(3):291-297.
[3]罗庆华. 中国大鲵营养成分研究进展及食品开发探讨[J]. 食品科学,2010,31(19):390-393.
[4]Shiina Y,Ii K,Iwanaga M. An Aeromonas veronii biovar sobria infection with disseminated intravascular gas production[J]. Journal of Infection and Chemotherapy,2004,10(1):37-41.
[5]刘爱华,鲁振省. 陕西省大鲵资源保护及管理初探[J]. 水利渔业,2007,27(4):69-71.
[6]问思恩,李 婵,侯淑敏. 陕西秦岭山区人工养殖大鲵肌肉中无机污染物含量分析[J]. 安徽农业科学,2011,39(16):9721-9722.
[7]孟 彦,杨焱清,张 燕,等. 野生和养殖大鲵群体遗传多样性的微卫星分析[J]. 生物多样性,2008,16(6):533-538.
[8]樊汶樵,孙志芳,杨 帆,等. 中国大鲵主要传染病病原研究进展[J]. 中国兽医杂志,2015,51(6):73-75.
[9]Amann R I,Ludwig W,Schleifer K H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation[J]. Microbiological Reviews,1995,59(1):143-169.
[10]李 莉,王錫昌,刘 源. 中国养殖大鲵的食用、药用价值及其开发利用研究进展[J]. 食品工业科技,2012,33(9):454-458.
[11]王 旭. 中国大鲵腐皮病病原学及组织病理学研究[D]. 雅安:四川农业大学,2011.
[12]吴中明,王 欢,敖弟书,等. 大鲵的迟钝爱德华菌感染[J]. 遵义医学院学报,2007,30(4):464-466.
[13]于 超. 羊胚胎胃肠道细菌分离与鉴定[D]. 长春:吉林农业大学,2012.
[14]杨 曼,兰阿峰,郭素芬,等. 免培养法研究野生川金丝猴肠道内生细菌多样性[J]. 微生物学通报,2014,41(8):1605-1612.
[15]刘 栋,兰阿峰,王 菲,等. 野生大鲵肠道细菌多样性及产酶活性研究[J]. 生物技术,2016,26(1):70-75,80.
[16]兰阿峰,杨 曼,郭素芬,等. 免培养法对大鲵肠道微生物多样性的研究[J]. 微生物学通报,2014,41(7):1342-1349.
[17]狄盼盼. 太湖养殖中华绒螯蟹肠道菌群多样性研究[D]. 上海:上海海洋大学,2013.
[18]李艳宇,任 盟,张丛尧,等. 红鳍东方鲀稚鱼肠道可培养细菌的多样性[J]. 水产科学,2015,34(10):652-656.
[19]孙振丽,宣引明,张 皓,等. 南美白对虾养殖环境及其肠道细菌多样性分析[J]. 中国水产科学,2016,23(3):594-605.
[20]刘蓓一. 扬州鹅肠道微生物多样性及其受饲粮纤维水平的调控研究[D]. 扬州:扬州大学,2012.
[21]马 晨,张和平,刘彩虹,等. 牛瘤胃与肠道微生物多样性的研究进展[J]. 动物营养学报,2014,26(4):852-862.
[22]孙 盛. 松辽黑猪肠道内容物及粪便总DNA提取方法对比研究及其盲肠微生物多样性分析[D]. 长春:吉林大学,2016.叶建生,赵素珍,徐宝伟,等. 不同养殖密度和水温对胭脂鱼生长的影响[J]. 江苏农业科学,2019,47(1):152-155.
