干细胞治疗产品质量管理策略分析
2019-08-08林巧
林巧
(苏州驾玉生物医药有限公司,江苏 苏州 215123)
1 细胞疗法发展背景
再生医学,特别是对干细胞的利用,是一种针对多种类型的身体损伤与病症的全新医疗形式。干细胞疗法是利用干细胞的自我更新能力和分化潜能,用于治疗血液系统疾病、神经系统疾病、心血管疾病、肝脏疾病和内分泌疾病等多种疾病。干细胞的发现及其在体内的作用是一项100多年来一直在被研究的课题。除了正在进行的大量的基础和临床研究外,科学家还在不断发现干细胞改善人类健康的新用途。
在这个漫长的路途中,世界各地的科学家们除了对这一神奇的细胞生物学有了更深入的了解之外,还对干细胞本身进行了深度研究,从植物到动物,再到人类病人,目的是为了寻求治疗疾病和损伤的新方法[1]。

表 1 干细胞研究领域发生的里程碑事件Table 1 Milestones in history of stem cell research
早期干细胞研究为研究人员提供了个性化用药的希望,因为它使科学家能够获得细胞中个体的独特基因组。然而,以脐带血为起始材料的干细胞治疗,因为经常牵涉到使用胎盘,曾引起西方相当一部分人包括以布什为代表的官员的反对,干细胞治疗领域一度陷入停滞。直到人类诱导多能干细胞(HiPSC)的研究成功实现了对脐带血依赖的脱离[3],各行相关人士才重新燃起了用iPSC来产生体内所有细胞类型的信心。继Shinya Yamanaka及其同事的开创性研究之后,iPSC的生成效率和稳定性得到不断提高,细胞研究尤其是基于干细胞功能开发的退化性疾病治疗又展现出非常光明的前景,能够治疗传统医学无法解决的疾病。为此,Shinya Yamanaka获得了2012年诺贝尔生理学或医学奖[3]。
干细胞治疗的产业化还处于起步阶段。目前为止,唯一一类得到美国FDA批准的干细胞治疗产品是从脐带血分离得到的异源性造血干细胞,其只能用于有自身造血功能缺陷、罹患血液系统癌症的病人身上。其他任何通过复杂细胞分化、转导、培养、有性能转变等的干细胞治疗产品目前均未获得FDA的上市许可。
第一个被FDA批准的胚胎干细胞临床试验是由美国加州的Geron公司申请的[4],用于恢复脊髓损伤后的神经再生功能。原定2010年启动临床试验,但由于在临床前研究中发现干细胞注射会引起囊肿的产生,FDA出于对安全性的考虑,没有让既定的临床实验进行下去。
目前,全球范围内有大量的干细胞临床试验正在进行,这些临床试验的适应证主要分布在眼科疾病、骨髓损伤和缺血性心脏病这3个领域(见表2)。
2 对人类细胞、组织以及细胞和组织产品的监管考虑
人类细胞、组织或基于细胞、组织的产品(FDA称之为“human cells, tissues”或“cellular or tissuebased products”,即HCT/Ps;欧盟称之为“advanced therapy medicinal products”,即ATMP)是一类复杂而多样化的治疗方法。不仅来源不同,且产品安全性因素亦不同,这类产品的制备过程也极其复杂,目前为止缺乏统一和成熟的产业化手段,再加上世界各国对这一类产品的研发和产业化的监管政策至今仍有很大的区别,如何有效控制干细胞治疗的质量是一个高难度的课题。鉴于此,医药监管机构、研究者、企业、医疗机构等必须将各自负责的产品环节,通过现代化的药品质量体系来分析,判断产品制备全过程的关键控制策略,以达到总体保证病人安全和干细胞产品有效性的最终目的。
HCT/Ps的实例包括但不限于来自外周血和脐带血的干/祖细胞、经处理的自体骨髓细胞、合成基质的上皮细胞以及精液或其他生殖组织。
以下项目不被认为是HCT/Ps:1)移植的血管化人体器官;2)分别列在美国联邦法规(CFR)21册(以下简称21 CFR)第607和207部分下的全血或血液成分或血液衍生物;3)分泌或提取的人类产品,如乳汁、胶原蛋白和细胞因子,但精液被认为是HCT/Ps;4)用于同源性用途的最低限度处理的骨髓,且不与其他物质结合(水、消毒剂或储存剂除外,但必须是该添加剂不会引起新的临床安全问题);5)用于制造HCT/Ps的辅助性产品;6)来源于人以外的动物的细胞、组织和器官;7)21 CFR 809.3 (a)中定义的体外诊断产品[5];8)42 CFR 121.2中所定义的使用器官修复的血管,用于器官移植并标记为“仅用于器官移植”[6]。
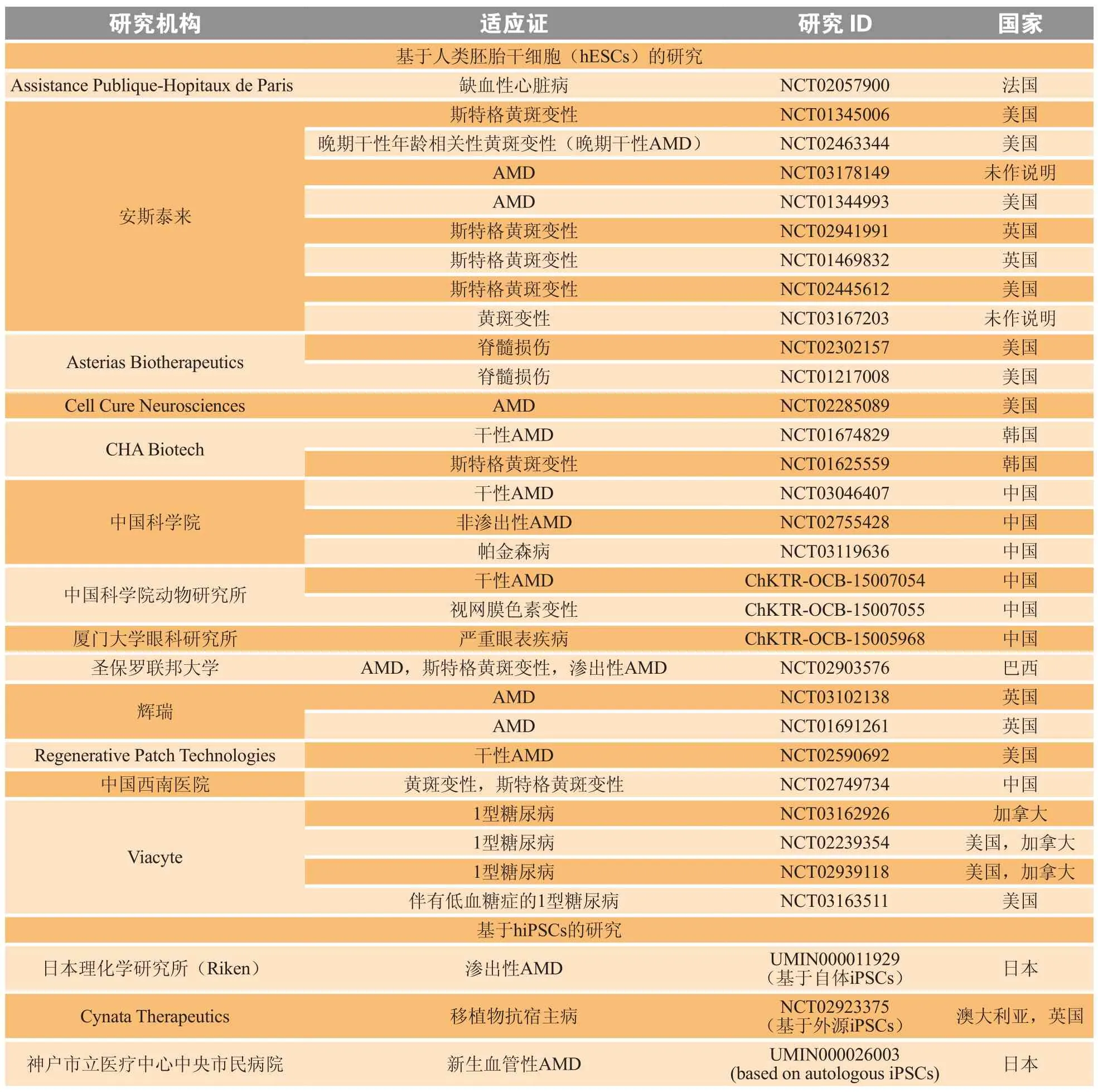
表 2 干细胞相关临床研究一览Table 2 Overview of stem cell related clinical research
2.1 美国FDA关于只经过最低程度处理或属于同源性HCT/Ps的现行条例
由于HCT/Ps的独特性质,FDA提出并于2005年实施了基于风险的分级HCT/Ps监管方法,并开始将《联邦食品、药物和化妆品法案》(FD&C法案)和《公共健康服务法案》(PHS法案)中的要求实施于符合药品、生物制品或医疗器械的产品。
根据PHS法案第361节(42U.S.C.264)中的传染病管理机构的规定,FDA通过联邦公告公布了一套被称为“组织规则”的规定。这些规定解释了不需要在上市前经过FDA批准的HCT/Ps类型,以及必须采取的登记产品、制造细节和报告步骤,以防止与HCT/Ps有关的潜在传染病的传入、传播和蔓延。在基于风险的方法制定分级时,该机构关注的公共卫生和监管问题包括:如何预防传染病的传播;需要采取哪些处理控制措施来防止可能出现的无效甚至有害产品的污染,并保持完整性和功能,从而使产品按预期工作;如何确保临床安全性和有效性。
2.2 美国FDA关于HCT/Ps作为生物药物的现行规定
FDA对于HCT/Ps监管采取了以风险控制为原则进行区分的2个途径:PHS法案361条管辖下的产品不需要通过FDA批准就可以上市,这些通常是风险较小或比较可控的产品。有关定义和监管政策收录在2017年11月FDA发布的对于相似体(homologous use)和最低程度处理(minimally manipulation)的细胞和组织治疗的监管指南中[7]。
此类规定可在21 CFR第1271部分找到[8]。在21 CFR 1271.10中,规定仅根据PHS法案第361节和21 CFR第1271部分确定了监管标准。如果HCT/Ps符合以下所有标准(21 CFR 1271.10(a)),则仅受PHS法案第361节和21 CFR第1271部分的监管:
1)HCT/Ps为最低程度处理产品,其中“最低程度处理”的定义如下:
● 对于结构组织,不会改变与用于重建、修复或替换有关的组织的原始相关特征的处理;
● 对于细胞或非结构组织,不会改变细胞或组织相关生物学特性的处理。
2)HCT/Ps仅被应用于同源的细胞或组织。
3)制造HCT/Ps过程不涉及细胞或组织与其他物品的组合,除了水、类晶体、消毒、防腐或储存剂之外,但前提是添加这些物质不会引起新的临床安全问题。
4)或者:
● HCT/Ps不具有全身效应,其主要功能不依赖于活细胞的代谢活动;
● HCT/Ps具有系统效应物,其主要功能依赖于活细胞的代谢活性,并且:①用于自体使用;②用于一级或二级血缘亲属异体使用;③用于生殖使用。
然而,没有涵盖到以上几点的HCT/Ps产品属于高风险产品,大多数细胞治疗药物包括干细胞治疗属于这一类。这些产品必须受FDA对于生物制品监管的PHS法案第351节和FD&C法案的约束,并要求获得FDA批准才能上市以及被商业化应用。此类HCT/Ps应符合的药物法规包括21 CFR第210和211部分中的要求,以及21 CFR第600至680部分中的适用要求。这类产品还同时受PHS法案第361条的管制,并须符合第1271部分的要求,这些要求主要针对防止污染物的传入、传播和蔓延。根据这些规定,细胞药物开发单位必须有合法注册公司来销售被批准的HCT/Ps(21CFR1271.1(b)(2))。
这些产品与其他生物药品一样,开发者/企业必须持有有效的生物制品上市许可证(PHS法案)(42 U.S.C. 262(a))才能合法在市场上销售。拿到上市许可证的前提是,FDA必须确定该生物制品符合适用要求并能确保其安全性、纯度和有效性,才可在获得产品上市批准的同时获得特定生物制品独有的编号和许可证(21 CFR 601.2(d))。对于临床研究,发起人必须根据FD&C法案(21U.S.C. 355(i))和FDA法规(21 CFR第312部分和21CFR 601.21)提出有效的临床试验申请(investigational new drug application,IND)。
1988年,FDA在21 CFR第312部分(子部分E)中发布了关于加速向病情严重的患者提供具有前景的治疗方法的规定[9]。该规定呼吁及早关注有希望治疗此类疾病的药物,包括及早与FDA就此类产品的申请进行磋商。在随后几年中,FD&C法案经多次修改,增加了几项新的加速产品开发和评审方案,包括快速通道选定、加速批准和突破性疗法资格。
2016年12月,美国国会修订了FD&C法案(21U.S.C.356)第506节,增加了新的第506(g)节,专门针对某些选定为“再生医学先进疗法”(regenerative medicine advanced therapy, RMAT)的产品作出加速开发和审查的规定。
用于治疗、改良、逆转或治愈严重疾病的RMAT,如果符合此类方案的标准,则有资格获得FDA的加速项目,包括快速通道、突破性疗法、RMAT、加速批准和优先审查。
2.3 欧盟有关干细胞治疗的最新法规
欧盟对于干细胞治疗的监管从2001年开始有快速发展[10-13]。到2014年,欧盟批准了第一个干细胞治疗产品Holoclar的上市申请。这是一款眼角膜上皮细胞干细胞治疗产品,可以用无创的方法治疗因化学物、强光照或炎症等促使角膜表面损伤而造成的疼痛、光过敏甚至失明[14]。
2018年5月,“晚期治疗药物产品生产管理规范准则”获欧盟通过。该准则指出,“应确保实施适当的制度,以确保ATMP及其起始和关键原材料的可追溯性”;“所使用的文件系统必须建立、控制、监测和记录所有可能直接或间接影响药品质量的活动,还应保存确保可追溯性所需的记录”,以及“对控制/缓解措施的风险和有效性的评估应以当前的科学知识和积累的经验为基础”。
2.4 中国有关干细胞治疗的监管政策和法规
中国对于细胞治疗,包括干细胞质量的监管虽然已有近10年历史,但比起欧美,无论是监管部门、职权分配、监管策略,还是法规条文以及对应的监管流程的执行,都处于一个比较初期和相对不稳定的状态。表3总结了中国最近几年与干细胞直接相关的法规条文。可见这些监管标准的出台,还没有到成熟稳定的阶段。此外,不同国家和地方的监管机构,也缺乏和谐、互补的监管流程,这种环境对于针对中国市场的干细胞研发和产业化企业带来了更多的困惑和挑战。

表 3 中国干细胞治疗相关监管规定Table 3 Overview of current regulations in China on stem cell therapy
3 干细胞治疗的质量管理
细胞治疗产品种类繁多,其生产工艺相对于化药,甚至比较成熟的单克隆抗体药物来说,更为复杂和多变。干细胞治疗作为一种全新的药物平台,在最近几年才有大量以产业化为目的的临床研究出现,由于具有技术含量高、生产过程复杂等特点,细胞疗法的安全性和疗效都需要经过复杂的确认过程,理论上应该完全按照生物药品来研发和监控。在药物的研发和产业化应用过程中,美国发展出了IND和上市申请[new drug application(NDA)/biologics license application(BLA)]的监管模式。在IND阶段的GLP+GMP+GCP制度,对药物的临床前安全性研究、药物生产的质量控制和临床试验的过程都进行了严格清晰的规范,保证了药物研发过程的高质量。而对于产业化药物的监管,保证了市场上产品的高质量及其安全性和有效性。
鉴于中国在干细胞治疗产品监管方面的现状,笔者提出以下要点,希望能帮助行业在研发和产业化过程中正确对待干细胞治疗的质量问题,采取ICHQ10、药物质量管理体系的理念和方法来执行,以达到保证产品安全性、纯度和有效性的目的。
3.1 高端生物医药产品的产品质量应为药品制造者的共同目标
在一个药物从研发到上市(市场应用)的过程中,以及上市后的商业化运作阶段,药物监管政策、法规的目的和宗旨是既能保护用药者得到安全有效的治疗,同时也督促这些研发和产业化机构能成功把科研成果转化为医药产品,使其在全国乃至全世界的市场上占有稳定和长远的商业竞争力。GLP+GMP+GCP等监管制度分别对于医药产品研发的不同阶段和方面设定相应的法规和指导原则,如药品的临床前安全性研究、药品生产的质量控制和药品临床试验的合规控制。所以,制药企业首先要对这些监管制度有充分的了解,建立与自己产品相匹配质量体系,才能保证其细胞治疗产品的初次研发到上市成功,而且以持续保证产品质量(安全性和有效性)来满足病人的需求并建立产品及其制造单位的信誉,最终达到商业化的成功。
3.2 干细胞质量管理的考量:安全性
任何药品的质量都包括它的安全性和有效性。在药品临床开发前期阶段,质量着重点是安全性。这个安全性必须在临床研究的全过程中保持稳定可靠,才能使产品有上市的可能性。对于治疗用的HiPSC细胞,有自体(autologous)和异体(allogeneic)两种,两者最大的差别在于异体细胞治疗可能产生免疫排斥性。自体干细胞由于是针对个人的,在制备的整个过程中必须严格保障患者(或正常人志愿者)的个人识别链条(chain of identity,COI),以及保证交付人完整记录和妥善保存所有生产环节的阶段和终产品(chain of custody),以免把他人的细胞输入受试者体内,引起强烈的免疫排斥性。
HiPSC的细胞库是高风险的关键原材料。细胞库管理不到位直接影响到干细胞治疗产品的关键工艺起点,因此建议必须严格按照GMP的要求来管理细胞库,这包括了冷藏仪器的验证、样品的标识、进出的流程等。
安全性的第2个要点是在整个治疗用干细胞的制备过程中不引入污染物。常见的污染来源有:1)医用容器及其他直接接触产品各阶段的包材;2)生产环境包括细胞存储容器和培养箱等;3)操作人员;4)原辅料;5)生产工艺中与产品有接触的仪器设备;6)在车间里同时生产的其他病人的细胞。由于细胞产品必须在促进细胞存活和生长的环境中进行,大多数细胞制剂的生产环境通常也有利于各种微生物的存活和扩张,只有在系统性地对以上因素进行全面分析和风险评估的基础上,采取有针对性的控制措施,才能避免有害微生物的污染,才能对细胞产品的安全性有持续的保障。
干细胞因其可塑性,理论上可能会被不同的外界因素激发而被诱入不同功能和不同分化程度的“命运”,可能产生的后果之一是细胞生长机制的失调而造成细胞癌变。所以,干细胞产品的致癌性是必须要控制的。
安全性的保障还包括对于在制备工艺过程中产生的有害物质有清晰的了解和严格的控制,包括杂质去除验证和分析方法检测等(分析方法在下文有单独阐述)。
3.3 干细胞质量管理的考量:有效性
干细胞治疗产品有效性的相关关键质量属性部分是对所有产品都适用的,例如对产品制备各步骤中的细胞活性必须要有控制策略及控制指标。对于不同的干细胞产品,在分化的过程中必须达到的特征表达,应该根据目的细胞特有的性能来控制。例如,用于重建心肌和治疗神经系统损伤的干细胞产品,必须要有除了细胞形态之外的功能性指标的质检策略。更具挑战性的是在细胞制备过程中相关质量属性由于细胞来源的个性化,即使在相同的工艺条件下,仍会发生非同步或非同频率的改变,这对于如何在工艺步骤中保持适当的控制,并证明制备的干细胞产品对目标适应证有效是一个巨大的考验。这些控制策略的不断成熟,必须在一个健全的质量体系内实现。
3.4 干细胞质量管理的考量:分析方法及质量控制
除了在研发及生产过程中系统化地采取质量管理措施来控制产品在安全性和有效性各方面的质量风险,对于原始志愿者[18]、其他物料、辅料、重要中间产品的质量控制均对分析方法提出了高度的要求。例如iPSC在激发、重编程、增殖、分化各步骤中需要用分析方法检测起始原料是否符合质量标准,工艺步骤是否达到既定的中间产品或最终产品的质量要求,直到终产品收获后的放行检测。由于此类细胞产品自身的高度可变性和不稳定性,分析方法必须要达到能够准确和精确地检测目的物质的要求,包括高风险的工艺杂质及因不稳定性产生的有害物质。也就是说,每个分析方法一定要经过验证来确定它是否符合测试的特殊目的及要求。这一切都必须在GMP体制的质量保证(QC)系统管理下才能实现,包括仪器设备和计算机系统的GMP管理。这些分析方法如果在产品临床开发过程中有所改变,必须对变更前后的影响作出评估,根据评估结果与药监机构进行必要的沟通并获得其认可。
4 结语
干细胞用于人类疾病的治疗已经有100多年的历史,医学科学家们在过去10年内的进步已排除了几个致命的困难,干细胞的潜力将在今后10年绽放成就和引领商业化的成功。干细胞治疗的最新进展包括哈佛大学与Sana Biotechnology签署的许可协议[19-20]。Sana Biotechnology是哈佛大学教职工最近共同创立的一家公司,专注于为患者创造和输送作为药物的工程细胞。哈佛大学的基础技术包括产生低免疫原性干细胞的方法,这些干细胞可以分化成任何类型的细胞,然后被移植到患者体内且不会引发免疫排斥反应。Sana Biotechnology打算制造低免疫原性的干细胞,并利用它们作为开发新的细胞疗法的起始材料。在适当的条件下,这些工程干细胞可以分化成不同类型的细胞,患者需要这些细胞来替代体内缺失或受损的细胞。相关研究显示,干细胞可以转化为心肌细胞(心肌)、内皮细胞(血管)、肝细胞(肝)或胰腺β细胞(产生胰岛素的细胞)。中国创新医药企业复星集团大踏步地进入干细胞行业,于2019年初宣布与英国干细胞治疗公司ReNeuron签署合作开发ReNeuron的2个临床干细胞治疗产品[21]。
在科学技术发展带来产业化的重大机会之际[22],干细胞治疗产品的生产及其质量管理,将成为下一个推动此类产品成功产业化的关键因素。对于复杂的干细胞治疗产品,只有遵循GMP管理的理念和现代化的质量体系,才能保证特定的细胞处理流程中的每一步都能够严格按照既定的程序和控制手段以及标准来进行。只有这样,细胞治疗产品在临床研究过程中才能充分体现工艺和产品质量与临床效果的相关性,以满足产品上市的生产和质量各方面的要求。
