转录组测序技术的研究和应用进展
2019-07-29崔凯吴伟伟刁其玉
崔凯 吴伟伟 刁其玉
(1. 中国农业科学院饲料研究所 农业部生物饲料重点实验室,北京 100081;2. 新疆畜牧科学院畜牧研究所,乌鲁木齐 830000)
转录组学是从整体转录水平系统研究基因转录图谱并揭示复杂生物学通路和性状调控网络分子机制的学科。在高通量测序技术发展以前,基于cDNA杂交荧光检测的高通量基因表达芯片(Expression array)和基因表达系列分析技术(Serial analysis of gene expression,SAGE)是从整体水平研究动植物组织中基因表达信息的主要手段。
在2008年左右,高通量测序技术开始应用于细胞和组织中转录本(主要是mRNA)的种类和表达量的研究,转录组测序(RNA sequencing,RNA-seq)这样的名词开始出现并被广泛应用[1]。与基因表达芯片方法不同,RNA-Seq不仅能够检测与现有基因组序列相对应的转录本,并能发现和定量新的转录本,对选择性剪接事件、新基因和转录本以及融合转录本的研究更具优势,从而能更加系统地研究转录组学。2010年前后,三代测序技术(单分子测序技术)兴起,因其具有测序读长较长的优点,在研究全长转录本上具有二代测序短reads所不能达到的优势。
随着测序技术的发展和成本的降低,使得核酸的检测与定量更加便捷和准确,高通量测序在转录组学的研究上越来越普遍,大有替代基因表达芯片的趋势。运用现有的转录组研究手段系统、准确地探究从DNA向RNA转录这一复杂而精细的调控层次,是揭示生物学过程中的复杂性状和解析转录调控网络的重要方面。
1 高通量测序平台
1.1 二代测序平台
20世纪70年代,第一代测序技术(Sanger双脱氧测序技术)的出现,实现了对核酸序列进行测序。但Sanger测序法的通量较低,不能满足大批量测序的要求,难以应用在组学测序的研究中。后来发展的高通量测序(High-throughput sequencing)技术,即二代测序(Next generation sequencing,NGS)技术,实现了测序的高通量和自动化,加速了转录组学研究的快速发展。目前,二代测序平台主要包括454 Life Sciences公司推出的454测序技术、Illumina公司和ABI公司相继推出的Solexa和SOLID 测序技术等,其中454测序技术平台最早实现商业化。在过去10年间,Illumina公司的Solexa技术,即边合成边测序(Sequencing by synthesis,SBS)技术发展迅速,其HiSeq系列的测序平台逐渐成为二代测序技术中最被广泛应用的平台。Illumina/Solexa的测序平台主要是采用边合成边测序(SBS)的方法,此种方法是将提取的核酸片段打断到几百bp大小后,加上接头和测序引物等序列,经PCR扩增后建成library,在含有接头序列的芯片(Flow cell)上对文库进行反应,每个反应循环中,标记4种荧光染料的碱基通过互补碱基配对被加入到单分子的合成中,这样通过CCD采集序列上的荧光信号,读取测序片段的碱基序列。Illumina现有桌面式和生产规模式两大类测序平台(表1),可以满足中小实验室以及大规模平台的二代测序需求。
1.2 三代测序平台
三代测序技术也叫单分子测序技术(Single molecule sequencing),具有超长读长(平均读长10-15 kb,最长读长可达60 kb)、无PCR扩增偏向性及GC偏好性的特点,被认为是进行全基因组de nove组装、全长转录本测序及表观遗传学测序的理想测序平台。PacBio公司的单分子实时测序技术(Single molecule real time sequencing,SMRT-seq)[2]和Oxford Nanopore Technologies的纳米孔单分子测序平台[3]是目前主流的三代测序平台。由于三代测序技术由于在测序时没有经过模板扩增,测序信号的检测相对于边合成边测序的二代测序技术较弱,容易在碱基识别时产生随机错误。因而PacBio平台的设计是对核酸分子进行环化测序(Circular consensus sequencing,CCS),反复读取reads后对碱基序列进行迭代校正,从而有效提高了单分子测序的准确性[2]。目前,PacBio 公司推出的 Sequel Ⅱ 测序平台,长序列reads的读取精确得到进一步提升,平均读长13.5 kb的长序列reads的读取精度可达99.8%[4]。

表1 几种主要测序平台特点
2 转录组测序
转录组测序(RNA-seq)就是利用高通量测序技术将细胞或组织中全部或部分mRNA、small RNA和no-codingRNA进行测序分析的技术。转录组测序和分析一般流程如图1所示。目前最常见的转录组测序是基于二代测序技术,以Illumina的NGS测序平台为主流。这种方法需要根据实验目的对RNA样本进行处理,将mRNA,miRNA,lncRNA其中的部分或全部反转录成cDNA文库,再利用高通量测序平台进行测序[1,5]。通常会根据测序对象长度的不同,在测序建库的时候会选择建立不同大小片段的文库。一般地,进行mRNA 测序,建库时通常建立几百bp大小片段的文库,选择双向测序较多;进行miRNA测序时,通常将miRNA进行分离,单独建立小片段文库后再进行单向测序;而长链非编码 RNA(long non-coding RNA,lncRNA)存在正向转录和反向转录,所以常采用链特异性建库测序。
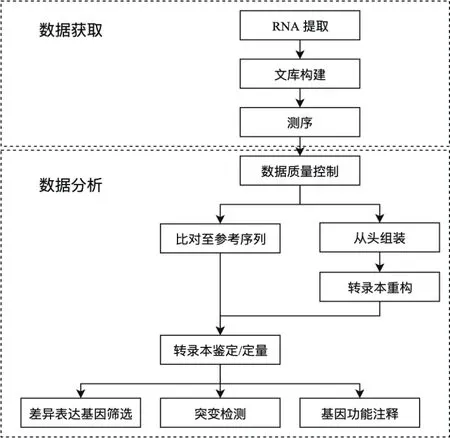
图1 转录组测序和分析流程示意图
2.1 mRNA测序
对于mRNA测序,利用mRNA在3′端具有poly-A的结构特点,富集出特定组织或细胞在特定时空条件下转录出来的不含内含子序列的mRNA分子,反转录成cDNA建库测序。根据测序得到的mRNA序列,可以精确地比对至参考基因组序列上,从而判断外显子与内含子的边界。对于无参考基因组的物种,通过对序列进行从头拼接(de noveassembly),得到转录本具体的序列信息。通过对不同物种、不同发育阶段的不同组织中的转录组进行研究,可以发现基因转录的物种特异性和时空差异的特点,为深入理解物种和性状的分子机理提供转录组水平的线索。
2.2 small RNA测序
Small RNA是指长度在20-50 nt的RNA分子,包括miRNA、siRNA、snoRNA和piRNA等,通过参与mRNA降解、抑制翻译过程、促进异染色质形成和DNA表观修饰等多种途径来调控生物学过程。根据small RNA 的5′端磷酸基和3′端羟基的结构特点,链接测序接头并筛选small RNA测序文库进行测序。miRNA在物种间的生物学功能较为保守,是small RNA测序研究中的重点。Hutvagner等[6]通过small RNA测序鉴定得到miRNA let-7,通过与mRNA不完全匹配发挥抑制翻译的作用。
2.3 lncRNA测序
长链非编码RNA(lncRNA)是一类长度在200 nt以上、无编码蛋白质功能的RNA分子,往往具有很强的物种、组织特异性。部分lncRNA位于基因的增强子区域,通过自身的转录而实现增强子的功能[7]。lncRNA调控方式多样且广泛存在于各类动植物细胞中,可以通过参与染色体结构形成以及与转录因子、蛋白质、RNA前体、miRNA结合等多种方式调节各类生物学分子的功能。部分lncRNA含有ploy-A尾结构,因而在mRNA的测序结果中往往包含部分lncRNA序列信息。目前对于lncRNA的研究,以寻找差异表达的lncRNA分子入手,主要依据lncRNA与关键编码基因的位置关系,进一步预测两者之间的调控关系。
1) 基于PID控制的参数为γ1=γ2=0.083,α1=α2=0.21,γ3=0.033,Kr=20。同时对PID控制器进行参数整定。Kp1=0.5,Kd1=0.002 5,Kd1=8;Kp2=1,Ki2=0.2,Kd2=0。
2.4 circRNA测序
环状RNA(circRNA)具有特殊的稳定性良好的成环结构,不容易被RNA酶降解,被认为在生物体内可以长效行使转录调控功能。同一段基因组序列可能会产生多种类型的circRNA分子,外显子和内含子的不同剪切组合使得circRNA可能包含多个外显子或内含子序列[8]。circRNA具有吸附miRNA分子的“海绵”作用,介入miRNA对mRNA的调控过程。circRNA对相同基因组位置上的mRNA转录有竞争性抑制作用[9],含有外显子的circRNA还可能开环并重新翻译[10]。由于circRNA在生物体内稳定地行使功能,并具有组织特异性表达模式,与宿主基因表达不太相关,被认为在作为相关疾病的临床诊断、预防的分子标志物以及药物治疗靶点等方面具有极大的潜力[11]。
2.5 全转录测序
mRNA的转录水平受lncRNA、small RNA和circRNA的调控作用影响,定量分析某个时空的细胞或特定组织中的生物分子网络和调控途径时,需要对整个转录组中全部的RNA分子进行定量和定性的研究。全转录组测序(Whole transcriptome sequencing)能够测定样本中的全部完整的转录本,主要包括mRNA和非编码RNA(lncRNA,circRNA和miRNA)。全转录本测序与常规RNA-seq的区别主要是建库方式的不同。全转录组测序在建库过程中需分别建立2个文库(mRNA+lncRNA+circRNA文库和miRNA文库)或3个文库(mRNA+lncRNA文库、circRNA文库和miRNA文库)。通过全转录组数据,不仅可以获得全部类型转录本的表达图谱,在此基础之上,对不同RNA分子进行鉴定和注释,分析其编码蛋白和调控功能,并对RNA分子之间的互作调控网络进行分析,从整体上全面系统的分析特定细胞在特定时空下的生物学特征。
2.6 单细胞转录组测序
Eberwine 等[12]和 Brady等[13]分别开发了通过体外转录线性扩增和PCR指数扩增单个细胞的互补DNA(cDNAs)技术,这类技术与高通量测序技术结合,衍生出了单个细胞内转录本测序技术(single cell RNA-seq,scRNA-seq)。单细胞转录组测序技术是在单细胞水平研究整个转录组的技术,用于评估单个细胞间基因表达的差异,能避免细胞类型混杂而引入的假阴性结果,有可能识别出无法通过混合细胞检测到的罕见的细胞群体。目前常见的单细胞测序平台包括Fluidigm、WaferGen、10×Genomics、和Illumina/Bio-Rad等测序平台。与其他RNA测序技术不同,scRNA-seq需要首先分离并获得单个细胞内的全部转录组。单细胞分离是scRNA-seq的关键步骤,主要通过连续稀释、显微操作分离、荧光激活细胞分选(Fluorescence-activated cell sorting,FACS)和微流控分离(Microfluidic technology)等技术实现。2009年,Tang等[14]首次报道利用scRNA-seq技术来鉴定早期发育阶段的不同类别细胞。
3 转录组测序文库的构建
进行转录组测序时,提取样本中总RNA后,去除rRNA,对目标RNA分子富集后进行测序文库的构建。测序文库,分为非链特异性文库(Non-strandspecific library)和链特异性文库(Strand-specific library)两种。非链特异性文库是指,RNA逆转录成双链cDNA,随机加上接头、不区分RNA的链的信息的文库。测序时以双链cDNA进行测序,无法区分mRNA的转录方向(图2-A)。链特异性文库可以分为2类,一种以化学修饰标记一条链,比如通过重硫酸盐处理RNA分子(图2-B),或者在第二链cDNA合成时引入dUTP(图2-C),然后降解含有U的链;另一种是以不同接头连接RNA分子或合成cDNA链的5′和3′末端,来区分正反义链(图2DH)。Levin等[15]对不同类型的链特性文库的复杂性、均匀性和覆盖连续性进行评价,并与已知基因组注释和表达谱的基因定量进行比较,再结合实验操作和计算的简便性,认为dUTP第二链标记的方法和Illumina RNA ligation方法效果较好。
在转录组测序时,区分RNA分子链的来源能够避免基因反义链上的reads的干扰,能够提高基因转录本鉴定和转录本定量的精确性。利用转录组数据进行从头拼接时,有助于划分转录本的边界,确定转录本的正义链信息。
4 转录组数据处理流程
转录本测序数据用于比较不同组别之间基因水平或转录本水平的定量差异时,其分析基本流程包括原始数据预处理、reads比对、转录本组装、新转录本预测、转录本表达水平分析等步骤。后续根据实验的目的,可进一步分析实验组与对照组之间的转录本表达差异情况,样本之间基因表达模式聚类,以及和其它组学数据进行联合分析等。其中,转录本组装是根据read比对的结果来识别样本中所有表达的转录本。如果没有可用的参考基因组序列,则可以直接使用从头组装方法执行此过程。一旦确定了所有转录本,就可以根据比对至转录本序列上的reads数目估计基因的表达丰度,进而在分组样本之间计算差异表达转录本并分析转录本表达情况与分组设计之间的生物学关联。
4.1 原始数据预处理
获得的二代测序原始数据后,需要对数据质量进行评估并进行质量控制(Quality control,QC),评估内容包括数据产出量、GC含量、rRNA含量、碱基质量分布和重复序列等。将其中低质量的reads和接头序列等去除,得到质控后的clean data用于后续分析。常用的质控软件有Trimmomatic[16]、RSeQC[17]、FASTX(http ://hannonlab.cshl.edu/fastx_toolkit/)、Trim Galore(https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/)等。以常用的Trimmomatic软件为例,对于双段测序的Illumina格式的rawdata,其默认处理参数为“ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW :4:15 MINLEN:36”,ILLUMINACLIP:TruSeq3-PE.fa:2:30:10含义是根据TruSeq3-PE.fa文件中的测序接头序列信息过滤原始数据中的测序接头和引物序列,允许最大错配碱基数为2,plindrome clip阈值为30,simple clip 阈值为10,根据这三个值的设定来判断接头序列与read的比对程度;LEADING:3,从序列开头去除碱基质量低于3的碱基;TRAILING:3,从序列结尾去除碱基质量低于3的碱基;SLIDINGWINDOW:4:15,设置4 bp窗口,碱基平均质量值阈值为15;MINLEN:36,若质控后read长度小于36 bp则丢弃此条read。QC后得到的数据称为clean data,用于后续的数据分析。
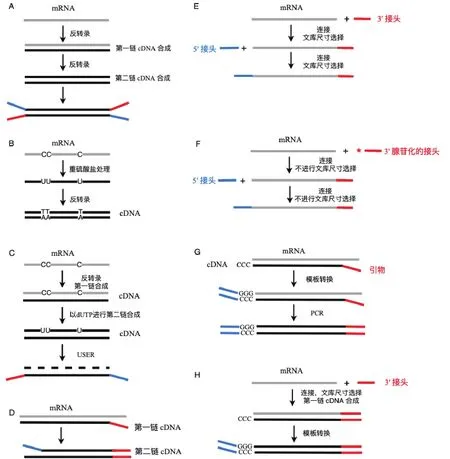
图2 几种类型文库的构建流程示意图
4.2 reads比对
转录组数据主要来自基因组的外显子序列,将测序获得的转录组reads比对至基因组序列上,会被内含子序列隔断。应用于转录组数据的比对软件, 常 用 的 有 bowtie[18],bowtie2[19],STAR[20],HISAT/HISAT2[21]等。这类软件适用于转录组reads的分割并比对至参考基因组序列。BWA[22]软件的比对算法被认为对于分割比对不敏感,因而不适合用于RNA序列与含有内含子序列的基因组序列之间的比对。
4.3 转录本组装
4.4 转录本预测
大多数基因有多种剪接形式,且有可能产生多种转录本,从而编码产生不同的蛋白,这样有可能造成一个基因有多种功能。对转录本测序数据进行拼接和组装后,不仅会得到已知的转录本信息,也会得到新的转录本序列,需要对新的转录本进行鉴定和注释,特别是之前研究较少的新的ncRNA转录本。
对于有参考基因组和转录本参考信息的物种,转录本结构主要是根据测序得到reads进行比对,reads覆盖了全部的转录本序列,依靠基因组序列组装出完整的转录本信息。对于无参考基因组的物种,需要自行组装出基因的转录本序列。得到的基因或转录本序列可以与同物种或近源物种的unigene和EST数据库进行比较,以判断得到的基因或转录本序列的可靠性。这一过程中常用blast方法进行比对,快速鉴定序列之间的相似度。在新的lncRNA的鉴定分析中,一般是根据lncRNA分子特性,在转录组数据中提取外显子总长>200 nt的转录本,然后根据开放阅读框预测和与已知的蛋白数据库进行比较,从而进一步将lncRNA从mRNA中分离出来。
4.5 转录本表达水平分析
将reads比对到相应的基因组位置或从头组装出转录本后,得到每个基因或转录本上的reads数在一定程度上可以反映其表达丰度。由于样本间的数据总产出量、样本间基因表达数目、样本内不同基因长度甚至是同一个基因内部不同转录本分布都可能存在明显差异,在不同样本之间对于同一个基因或转录本的表达量进行比较时,则需要对于样本间的数据进行归一化处理[29]。处理后的转录本表达量一般以RPKM(Reads per kilobase per million mapped reads)、FPKM(Fragments per kilobase per million mapped reads) 或 TPM(Transcripts per million) 这3类数值表示。RPKM针对早期的SE测序,FPKM则是在PE测序上对RPKM的校正,且应用于双段测序的RNA-seq分析中。对于单端测序结果,基因的RPKM与FPKM值相等。TPM以转录本条数代表每个转录本的表达量,当样本之间表达的基因综述差异很大的时候,TPM值比FPKM值更能代表转录本的表达量。Cufflinks[30]、DESeq/DESeq2[31-32]、EDGR[33]等软件可用来进行表达量的确定。另外在进行不同样本之间的基因表达量比较分析时,依据实验设计利用统计学方法检验实验组与对照组之间的差异基因。由于同时对成千上万的基因进行统计检验,因而需要考虑多重检验引入的假阳性升高,因而常用FDR等多重检验校正的方法对比较分析的显著性进行校正。
scRNA-seq中基因表达量确定与常规RNA-seq数据处理思路一致,但由于scRNA-seq的产出数据中一般噪音较大,并且有大量的空值(Dropouts),在基因表达矩阵中超过50%的基因为空值也很常见,因而需要使用填充算法进行数据的修正[34-35]。对于scRNA-seq空值的解释,除部分基因本身在该单个细胞中不表达,还可能是因为一些基因表达量较低或者是测序深度不够,导致在实验中未被检出[34]。在数据分析中,需要谨慎对待空值,根据细胞-细胞、基因-基因之间关系对空值进行推断。推断scRNA-seq中的技术噪声比较困难,单个细胞之间的测序结果属于生物学重复而不能用于推断技术重复偏差。
4.6 变异检测
分析转录组测序数据,可以获得样本的全部转录本的序列信息,包括转录本上全部的SNP和Indel等突变类型。转录生成的RNA分子在翻译之前可能会经历多种修饰[36],如A-to-I的RNA编辑过程,从而进一步增加转录组的复杂性。分析转录本中的突变信息,可以捕获基因从DNA向RNA转录中修饰过程,从而探究转录过程中复杂的调控机制。SAMtools[37]、BCFtools[38]和 GATK[39]等软件可用来检测转录组中相关的变异。
5 RNA-seq的应用
5.1 确定基因的表达模式
RNA-seq技术使得研究整个转录组中基因表达情况更为快速和容易,对于有参考基因组的物种或无参考基因组的物种,采取不同的分析策略,即可高效地获得某一特定状态下细胞或组织中的全部转录本信息,进而研究不同组织器官或发育阶段之间的基因表达模式差异。另外,随着转录组测序数据的积累,从而建立起各类转录组数据库,这些信息的积累为后续进一步研究特定生理条件下的转录本种类和表达量、转录本之间互作以及转录调控研究等提供参考。例如,基于RNA-seq数据建立起来的拟南芥的基因表达谱数据库TraVA(http://travadb.org/),包含了79个样本的25 706个蛋白质编码基因,可以提供不同器官和发育阶段之间的基因表达谱和差异表达基因的研究[40]。基于miRNA-seq建立的miRBase数据库收录了200多个物种的数万条miRNA信息,可用于进行miRNA注释和比较[41]。
5.2 用于发现新转录本
基于NGS技术的转录组测序,通过拼接算法可以获得完整的转录本序列,但对于存在多个可变剪接形式的基因,不同转录本之间的识别仍然存在困难。单分子测序技术应用于转录组测序,可以用测序读长较长的优势直接获得完整的转录本序列,不再需要对于NGS短的reads进行组装,可以准确获得不同可变剪接的转录本序列和表达量[42]。Wang等[43]利用 Pacbio Isoform Sequence(Iso-seq)平台分析鉴定玉米和高粱的异构体,发现超过40%的转录本具有新颖和多种多样的剪接方式。
5.3 非编码RNA的调控机制
非编码RNA不能形成蛋白产物,但其在生物学过程中发挥着不可忽略的重要的调控功能。近年来,通过NGS技术和RNA-seq在各种细胞和组织中发现了大量的ncRNA,它们的类型和表达量,往往反映了物种特征以及特定生理状态。特别在动植物育种、抗病和环境适应性的研究中,ncRNA的作用越来越受到重视。在拟南芥中,病原菌侵染过程中可以激发一些小RNA,在相应的通路中发挥作用,最终增强植物抗病性。Zhao 等[44]通过对拟南芥进行转录组的测序,研究lncRNA表达谱,从鉴定出的6 510个lncRNA中进一步分析了影响开花的lncRNA及其作用机理。Ré等[45]通过对低温环境中的拟南芥种子进行miRNA测序,鉴定出低温状态下独立于HYL1/SE产生的特定miRNA,分析其与低温环境适应性的联系。
5.4 单细胞转录组
从单个细胞水平上进行转录本研究,能够深入分析细胞之间的异质性,从而解析不同类型细胞的生理特点。对于特定组织研究其功能,必须要了解区分其细胞类型组成,不同的细胞类型可能发挥了特异的生理功能。scRNA-seq成为深入研究不同类型细胞的转录本的差异以及细胞之间的组合的解决方案。目前已经有多种方法能够实现单细胞分离并进行scRNA-seq的研究并被大量的应用在肿瘤细胞的转录组研究中[46-47]。Moffitt等[48]利用 scRNA-seq技术对大脑中的不同类型细胞进行鉴定,不仅发现了之前未知的神经元亚型,结合MERFISH这样的空间成像技术,还允许对鉴定出来的不同类型细胞的空间分布进行研究,明确不同的基因在不同类型的细胞中表达差异。
6 展望
转录组测序技术能够确定信使RNA(mRNA)和非编码RNAs(ncRNA)序列和转录基因的结构,并定量的动态表达每个转录本在不同生物学下的模式条件。随着测序技术的进一步发展,为转录组的定位和定量的研究提供了多种新的解决方法。总体来说,目前的RNA-seq仍然以基于二代测序为主,基于三代测序技术进行RNA-seq成为转录组学研究的一个重要方向。目前的高通量测序技术都需经过建库过程,即首先将大量的RNA分子富集后,转化为cDNA测序文库,并在建库过程中对cDNA分子的片段连接上测序引物接头和样本标记序列。应用最广泛的Illumina二代测序平台,采用边合成边测序的策略,对建库后的分子进行双端测序或单端测序。PacBio的单分子实时测序技术,具有读长较长的优点,能够进行全长转录组的研究,特别适合用于发现新转录本。根据实验目的和研究对象,基于现有的测序平台,已经可以在整体水平系统地开展多种RNA分子的研究,除了全转录组和全长转录本的研究外,单细胞转录组的研究是近来的研究热点。随着单细胞分离以及单分子测序技术的发展,单细胞转录组测序技术在异质性细胞的转录组研究中具有广阔的前景。RNA-seq技术的发展为从整体转录水平,细致精确地研究转录调控网络与性状之间的关系提供了有效手段。
