同核铁系双金属络合物及其在均相催化体系中的应用
2019-07-26王露孙威刘超
王露 ,孙威 ,2,刘超 ,*
1中国科学院兰州化学物理研究所,羰基合成与选择氧化国家重点实验室,兰州 7300002中国科学院大学,北京 100049

1 引言
在现代有机合成化学中,金属催化剂具有非常重要的作用。金属催化剂的种类繁多,活性区别很大。随着合成化学的不断发展,人们正在尝试开发更多高活性的催化剂来活化一些惰性化学键或者惰性有机分子。在不断的探索中,人们发现双核金属络合物会展现出不同于单核金属催化剂的催化活性1,2。在双核过渡金属催化体系中,因两个金属中心存在协同作用而表现出了较好的催化活性3-6。基于双核金属络合物作为催化剂的催化体系研究目前也取得了一定的进展。其中Fe、Co、Ni为第四周期第VIII族元素,统称为“铁系元素”,具有储量丰富、廉价易得、相对安全无毒、环境友好、生物兼容性好等特点。其次,铁系金属的金属盐及其络合物在空气中大多能稳定存在,且大部分可直接购买。而且Fe、Co、Ni既可以实现双电子转移过程也可以实现单电子转移过程,这些特性能够使得催化剂可以催化多种类型的反应,因此引起了人们的广泛关注。目前,已有较多文献对双核Fe、Co、Ni金属络合物的合成及其性质进行了报道7-9,因此本综述主要集中在同核双核铁系金属络合物在均相催化中的应用,其中双核羰基金属络合物不在此综述范围内。
2 双核Fe络合物催化的化学转化
在2008年,Tatsumi课题组10报道了N2桥连的NHC配位的双核铁络合物1a (图1),每一个铁原子都是典型的三脚钢琴凳的构型,然后通过N2的弱配位相连接,同时该络合物是具有抗磁性的。实验结果表明该络合物可以活化杂环的C——H键,但是目前还不能实现其催化反应。2017年,山东大学王文广课题组11报道了一例与其结构类似的双核铁络合物[Cp*(Ph2PC6H4S)Fe]2(μ-N2) (图1),该化合物1b是一种中心对称的结构,两个Cp*Fe(Ph2PC6H4S)结构由直线型的N≡N连接,Fe——N——N的键角为177.3(4)°。通过单晶衍射、拉曼、红外手段对其结构进行了表征,两个铁原子中心通过N2的弱配位相连接。

图1 双核铁络合物1a 10和1b 11的结构比较Fig. 1 Comparison of the structure of dinuclear iron complexes 1a 10 and 1b 11.
随后作者考察了络合物1b对N-杂芳环的硼氢化反应的活性。结果表明该络合物对含氮杂环类化合物的硼氢化反应具有很高的活性,可以在50 °C甚至室温下高产率高选择性地得到N-硼化的产物(图2)。该络合物的发现为杂环化合物上引入硼基团提供了一种新的合成方法与合成思路。随后,作者对该反应机理也做出了较为详细的研究(图3)。在该反应过程中,络合物1b首先会与杂环的N原子配位生成中间体1c,而后HBpin上的B与S原子配位生成中间体1d,接着发生H原子的转移过程,最终生成目标产物。2013年,Chirik课题组12发现了以双亚胺吡啶(PDI型)为配体的,N2连接的双核铁络合物在催化烯烃硼氢化反应中具有一定的活性。但是作者在研究中发现,使用PDI型配体配位的铁络合物,无论单核还是双核金属催化剂,催化活性相差不大。
Betley课题组13在2016年报道了由两个氯桥连的双核铁络合物[(tBuL)FeCl]2(1e)。该络合物是一种绿色的微晶粉末,可以与芳基叠氮化合物反应,生成两种新的高自旋Fe(III)络合物。一种是单核Fe-N络合物1f;另外一种是以氮桥连的双核铁络合物1g。络合物1e可以活化烯烃α-位的C——H键和甲苯苄位C——H键,实现C——N成键反应,以及苯乙烯类化合物与芳基叠氮化合物生成氮杂环丙烷的转化(图4)。

图2 络合物1b催化含氮杂环的硼氢化反应11Fig. 2 1,2-hydroboration of N-heterocycles catalyzed by complex 1b 11.

图3 反应中间体以及H原子的转移过程11Fig. 3 Intermediate of the reaction and H atom transfer process 11.

图4 络合物1e催化的C——N成键反应13Fig. 4 C——N bond formation catalyzed by complex 1e 13.
在对机理的研究过程中,作者猜测化合物1g可能是反应的主要中间体,并且通过19F NMR监测催化剂之间的相互转化(图5)。以芳基叠氮化合物与环己烯偶联反应为例说明,首先催化剂1e与芳基叠氮化合物反应生成中间体1f和1g,1f与芳基叠氮化合物在催化剂1e的作用下也会在一定程度上转化成1g,1g会直接与C-H化合物发生氢原子的攫取生成1h以及环己烯自由基,1h与环己烯自由基相互作用生成目标产物,同时也会生成1e。

图5 络合物1e催化C——N成键的反应机理13Fig. 5 Mechanism of C——N bond formation catalyzed by complex 1e 13.
2015年,曲景平课题组14报道了以硫负离子桥连、腈配位的双核Fe络合物(1i)催化腈的水合反应制备酰胺。在工业生产中,腈的水合反应都需要在强酸或者强碱的条件下完成,条件苛刻,会产生很多废物,不符合绿色化学的理念。早在1994年时,Que课题组15就报道了一例以氧原子连接的双核Fe促进腈类化合物水解成酰胺,但是生成的酰胺的N、O原子分别与双核Fe配位,形成稳定的五元环,无法得到游离的酰胺。曲景平教授通过实验研究发现,当向该反应体系中加入HBF4·Et2O后,可以使其上述与双核铁配位的酰胺解离出来从而得到游离的酰胺(图6)。该反应体系可以兼容烷基腈、烯丙基腈、N-氰基类以及芳香腈类化合物,且可以在室温下高产率转化成相应的酰胺。
偶联反应在有机合成中有着非常广泛的应用,一般都是用活性较高的芳基或者是烯基卤代物(Cl、Br、I)做为亲电试剂。2012年,邓亮课题组16实现了双核铁络合物催化的非活化一级烷基氟代物与芳基格氏试剂的偶联反应(图7)。作者通过与单核铁催化剂相比,如Fe(acac)3、(IPr2Me2)Fe(Mes)2等都几乎得不到目标产物。当使用双核铁催化剂(Cat.Fe2)时,却能高效高选择性的得到目标产物,反应体系可以兼容各种官能团取代的芳基格氏试剂以及烷基氟代物。随后的反应机理研究表明该反应很可能是一个自由基过程(图7)。首先,催化剂前体Cat.Fe2在格氏试剂的作用下发生配体交换,生成低配位的负离子1j。而后1j与RF作用,发生单电子转移,产生烷基自由基以及与铁配位的芳基自由基,最后这两种自由基偶联后得到目标产物。
3 双核Co络合物及其催化的化学转化

图6 络合物1i催化的腈水解成酰胺机理15Fig. 6 Mechanism of hydration of nitrile to amide catalyzed by complex 1i 15.

图7 络合物1j催化的芳基格氏试剂与一级烷基氟代物偶联反应及其机理16Fig. 7 Complex 1j catalyzed cross-coupling of aryl Grignard reagents with primary alkyl fluorides and the proposed mechanism 16.
从目前的文献报道来看,双核Co络合物在不对称加成以及CO2催化转化方面应用的较多。

图8 络合物(R)-(OAc)2-2a催化的β-酮酸酯与炔酮的不对称1,4-加成反应17Fig. 8 Complex (R)-(OAc)2-2a catalyzed asymmetric 1,4-addition of β-keto ester to alkynone 17.
在2009年,Shibasaki课题组17报道了席夫碱骨架的双核Co(III)络合物(R)-(OAc)2-2a催化的β-酮酸酯与炔酮的不对称1,4-加成反应(图8)。该反应的产率和ee (对映体过量)值都可以达到95%以上。这种骨架的双核Co(III)络合物在空气中是稳定的,存放六个月以上反应活性都不会降低。而且在该催化体系中,催化剂的当量(即用量)可以降低至0.1%。作者通过与单核Co金属催化剂的对照,发现使用单核Co催化剂时,选择性和产率都会大大降低,甚至根本不能得到目标产物。作者认为两个Co中心的相互作用对该反应起着很大的作用,目前具体作用还不清楚。
在2005年,Gao课题组18合成了席夫碱骨架的Robson类型大环配体的双核Co(II)和Co(III)络合物,用于苯乙烯与重氮乙酸乙酯的不对称环丙烷化反应(图9)。在研究过程中,作者发现,两种配体L1和L2组成的二价和三价L1-Co(II)、L1-Co(III)、L2-Co(II)、L2-Co(III)钴络合物,在催化该反应时,产率、ee值以及反式/顺式环丙烷的比例都很接近,说明这几种络合物催化该反应的机理是一样的。在该反应过程中,会产生两个金属-卡宾中间体。由于这两个卡宾会相互作用,导致这两个卡宾重排,成平行的位置,而这两个金属卡宾与络合物的平面呈垂直状态。这样的位置降低酯之间的空间位阻,以及与轴向氢原子间的相互作用。
过渡金属催化的CO2与环氧乙烷类化合物制备环状碳酸酯以及聚碳酸酯是非常重要的一类反应。该类反应能有效的利用CO2合成大分子量且可以生物降解的聚合物(图10)。Williams课题组19发展了两种双核钴(Co(II/II)和Co(II/III)络合物,在1个大气压,80 °C的条件下完成了环氧乙烷与CO2的聚合反应。当使用络合物2f时,TON (turnover number,单位活性位转化的底物分子数)可以达到420,TOF (turnover frequency,单位时间单位活化位转化的底物分子数)达到159 h-1,而分子量最高可以达到6300。但是该反应还存在一定的缺陷,聚合物分子量比较小,可能是由于链转移反应的影响。

图9 双核Co催化的环丙烷化反应18Fig. 9 Dimetallic Co catalyzed cyclopropanation 18.
在以往的催化体系中,都会观测到环氧乙烷自身聚合的副产物,且分子量也比较小。为了解决这一问题,Ko课题组20于2016年合成了以双苯并三唑亚胺酚类为基本骨架的双核Ni、Co络合物,并将其应用到环己烯氧化物与CO2的反应中(图11)。研究发现双核Co络合物(2g)以及双核Ni(2h)展现出很好的催化活性,聚合的选择性可以达到99%以上。特别是络合物2h,24 h时TON > 1100,聚甲基乙撑碳酸酯(PCHC)/环己烯碳酸酯(CHC)的选择性也在99/1以上,且催化剂的当量可以降低至0.03125%。当反应温度升高至150 °C时,催化剂2h仍然可以稳定存在,并且保持很高的反应活性。当具有相同配体骨架的单核Ni催化剂2j时,该聚合反应基本不进行。当Bu4NX (X = Cl,Br,I)作为共催化剂时,反应的选择性完全发生改变,环己烯碳酸酯(CHC)成为主产物,双核Co (2g)的活性最高,选择性在99%以上,几乎观测不到聚合的产物生成。
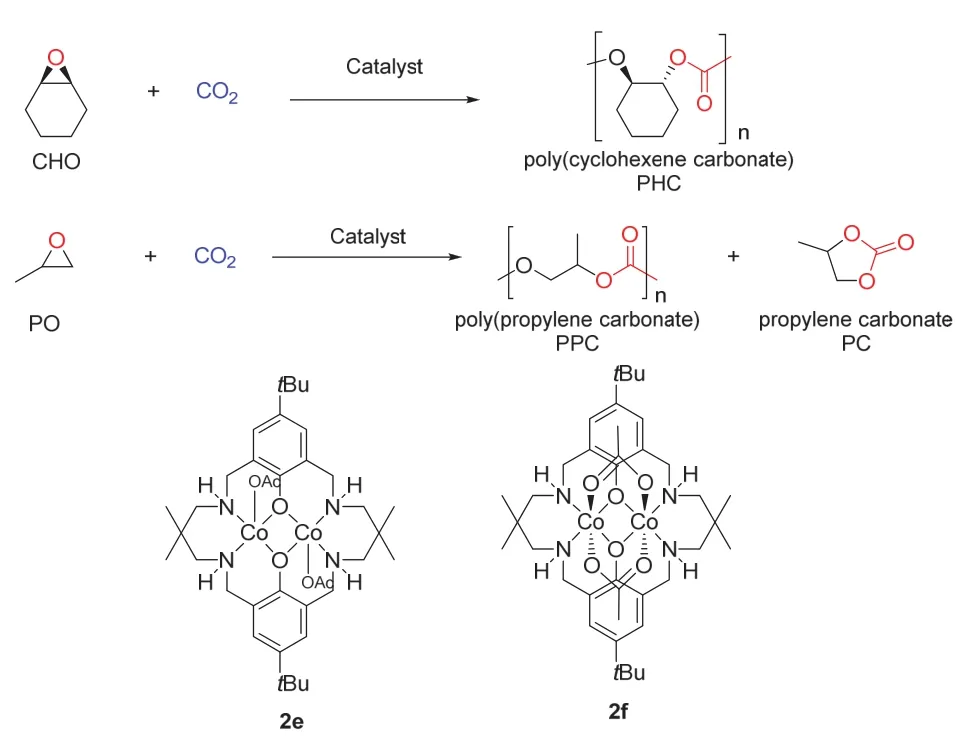
图10 双核Co催化的环氧乙烷类化合物与CO2的聚合反应19Fig. 10 Copolymerization of CO2 with ethylene oxide compounds catalyzed by dimetallic Co 19.

图11 双核Co催化的环己烯氧化物与CO2的聚合20Fig. 11 Copolymerization of cyclohexene oxide and CO2 catalyzed by dimetallic Co 20.
二胺类型(DiBTP)的骨架比亚胺类型(BiIBTP)骨架的灵活性更强,2016年Lin课题组21报道了DiBTP类型的双核Ni络合物(图12)对环己烯氧化物(CHO)与CO2的共聚具有很好的催化活性,TON值可以达到4000以上,TOF值也可以到400 h-1。同时,这类催化剂还可以催化乙烯基取代的环己烯氧化物与CO2的聚合反应,且分子量可以达到41800。
4 双核Ni络合物及其催化的化学转化
基于席夫碱骨架的双核镍络合物是一种很稳定的络合物,该催化剂在不对称催化反应中有着广泛的应用。

图12 DiBTP类型的双核Ni、Co络合物21Fig. 12 Bimetallic Ni and Co complexes containing the DiBTP ligand backbone 21.
2008年,Shibasaki和Matsunaga课题组22报道了基于席夫碱骨架的双核Ni络合物(R)-Ni2-3c催化的不对称Mannich加成反应合成具有手性的α,β-二氨基羧酸衍生物(图13)。这种双核Ni络合物可以在空气中稳定存放3个月,且催化活性不会降低,当使用3a、3b作为反应底物时,能以95%的收率,91 : 9的dr(非对映比)值以及98%的ee值得到目标产物。该金属催化剂与席夫碱骨架的单核Ni络合物的反应活性相差较大。当使用单核Ni作为催化剂时,反应活性、产率和选择性都有所降低。由此说明,两个金属Ni中心的相互作用对反应选择性以及产率都有较大的影响,目前该作用还不清楚,还在进一步的研究中。
同年,该课题组使用β-酮磷酸酯时,也能获得理想的产率与选择性(图14,方程式1)23。作者猜测这种双核金属Ni-芳基氧的部分是起到Brønsted碱的作用,β-酮磷酸酯烯醇式中的氧原子以及磷酸酯上的氧与其中一个Ni中心配位,N-Boc亚胺中的羰基氧会与另一个Ni中心配位(图14)。最终以90%产率,20 : 1的dr值,99%的ee值得到反式加成的产物。随后,他们继续使用该催化剂在室温下完成了亚胺或者硝基取代的烯烃类化合物对α,β-不饱和-γ-丁内酰胺的不对称加成反应(图14,方程式2-3),dr值可以达到30 : 1,ee值高达99%,而以席夫碱为基本骨架的单核Ni金属催化剂是没有活性的,即使反应时间延长至24 h也检测不到目标产物24。

图14 络合物(R)-Ni2-3c催化的不对称加成以及反应中间体的猜测23,24Fig. 14 Complex (R)-Ni2-3c catalytic asymmetric addition reaction and the proposed intermediate 23,24.

图13 双核Ni与单核Ni催化剂的催化活性对比22Fig. 13 Comparison the catalytic activity of bimetallic Ni with mononuclear Ni 22.
该课题组发现该催化剂对于β-酮酸酯与甲醛的不对称Aldol缩合反应也具有很高催化活性25,其中催化剂的当量可以降低至0.1%,在较温和的条件下最高可以达到95%的ee值。除此之外,该类催化剂还被他们用于催化1,1-二磷酸酯烯烃的不对称1,4-加成反应26。随后,他们将这类催化剂用于α-酮酰胺与不饱和β-酮酸酯和硝基化合物的不对称反应27,28。他们在文章中提出,α-酮酰胺与不饱和β-酮酸酯先形成烯醇结构,双核镍催化剂中的两个金属中心分别与两种底物作用,一个Ni中心与烯醇结构配位,另一个Ni中心作为Lewis酸与硝基取代的烯烃配位,最后两分子发生加成-质子化作用得到目标产物。
2012年,Shibasaki课题组29报道了该结构的双核镍催化剂还可以催化3-取代戊二酸酐的开环酯化反应,在较温和的条件下就得到γ-酸酯类化合物(图15)。当使用S构型的催化剂前体(S)-Ni2-3c时,得到的目标产物也会发生构型翻转,ee值保持不变。
2017年,周辉课题组30报道了含有磺酰胺结构的NON配位的双核Ni (3g)催化的不对称曼尼希反应(图16)。该反应的催化剂当量可以降低至0.05%,而产率和ee值基本上不会发生改变,反应可以扩大至4 g以上。

图15 络合物(R)-Ni2-3c催化3-取代戊二酸酐的去不对称化反应29Fig. 15 The desymmetrization of 3-substituted glutaric anhydrides catalyzed by complex (R)-Ni2-3c 29.

图16 络合物3g催化的不对称曼尼希反应30Fig. 16 The asymmetric Mannich reaction catalyzed by complex 3g 30.
除了席夫碱骨架的双核Ni具有很好的催化活性外,NHC配位的具有Ni——Ni键的双核Ni络合物在催化化学转化时也具有很好的活性。2003年,Sigman课题组31报道了NHC配位的单齿π-烯丙基Ni(II)络合物催化对氧气具有一定的活化能力。该络合物是Ni(COD)2与烯丙基类化合物(烯丙基氯或者肉桂基氯)、卡宾配体合成的π-烯丙基Ni(II)-NHC,该络合物可以被氧气氧化成O-桥连的Ni(II)络合物。但是当使用Ni(COD)2与4-氯-2,4-二甲基-2-戊烯反应时,却得到了一些混合物,最终以10%-20%的收率分离得到了单齿卡宾配位的具有Ni——Ni键的氯桥联的双核Ni(I)络合物,这种中性单齿配体配位的Ni络合物是比较少见的。接着作者通过Ni(COD)2与NiCl2(DME)在卡宾配体的作用下,可以以很高产率得到这种络合物(图17)。
2009年,Hillhouse课题组32报道了与该结构类似的双核Ni络合物,[(IPr)Ni(μ-Cl)]2(3h)。[(IPr)Ni(μ-Cl)]2与2,4,6-三甲基苯的叠氮化合物反应生成d8-d8酰亚胺Ni的络合物[(IPr)NiCl]2(μ-NMes) (3i),使用NaBArF攫取氯原子后得到d8-d8二聚体[{(IPr)Ni}2(μ-Cl)(μ-NMes)]BArF4(3k)。{(IPr)NiCl}2(μ-NMes)可以被KC8还原成d8-d9混合价态的{(IPr)Ni}2(μ-Cl)(μ-NMes) (3l)络合物。{(IPr)NiCl}2(μ-NMes) (3i)可以发生氮烯转移到CO,PMe3以及CNR (R = PhCH2,CMe3)的过程(图18)。最终,Hillhouse 课 题 组 把 {(IPr)Ni(μ-Cl)}2(3h)以 及[{(IPr)Ni}2(μ-Cl)(μ-NMes)]BArF4(3k)用于催化有机叠氮化合物的氮烯转化合成一系列的碳化二亚胺、异氰酸酯类的反应中(图19)。
这类以卡宾为配体的,卤素桥连的双核金属Ni催化剂在催化偶联反应中也具有很重要的应用价值,较为常见的应用有Kumada-Tamao-Corrious偶联33,34,Buchwald-Hartwig胺化35,Suzuki-Miyaura偶联36以及芳基碘代物的三氟甲硒基化反应37(图20)。

图17 氯桥联的双核Ni络合物的合成31Fig. 17 Synthesis of bimetallic Ni complexes 31.

图18 {(NHC)Ni(μ-Cl)}2络合物的转化32Fig. 18 The transformation of complex{(NHC)Ni(μ-Cl)}2 32.

图19 双核Ni催化的有机叠氮化合物的转化32Fig. 19 The conversion of organoazides catalyzed by bimetallic Ni complex 32.
除了以卡宾为配体的双核金属Ni可以催化偶联反应外,2014年,王中夏课题组38报道了N配体的单核Ni与双核Ni催化的芳基卤代物(拟卤代物)与锌试剂或者格氏试剂的偶联反应(图21)。通过底物扩展以及与单核Ni催化剂的对比发现,双核Ni催化剂的兼容性更好,反应活性也更高一些,有的几乎能得到当量转化的产物。主要是由于双核金属络合物的两个金属中心分别会与芳基卤代物的芳基和C——X键作用,这种协同作用有利于金属与芳基卤代物的氧化加成。
2014年,Uyeda课题组39合成了一种基于1,8-萘啶-二亚胺结构(NDI)的双核Ni络合物(iPrNDI)Ni2(C6H6) (3n)。该催化剂可以发生多电子的氧化还原过程,有五种不同的氧化态(图22),可以催化许多类型的化学转化。
由于该双核金属Ni络合物具有多种氧化还原态,Uyeda课题组将其应用于各种催化反应中。2015年,Uyeda课题组40发现该金属催化剂可以催化烯烃、炔烃、醛、酮、酰胺等化合物的硅氢化反应,都能以很高产率得到目标产物(图23)。随后也与单核金属Ni催化进行了比较,发现催化剂3n比单核催化剂3o和3p的效率高很多,3o和3p几乎不能催化二苯乙炔的硅氢化反应。在反应过程中,该双核金属Ni络合物可以很快的与Ph2SiH2、Et2SiH2反应生成络合物。通过单晶衍射,发现在该配位过程中,H——Si——H的键角以及Si——H键的键长都发生了较大的改变。而NDI中的π-电子有利于该过程的进行。

图20 以卡宾为配体的双核Ni催化的偶联反应33-37Fig. 20 cross-coupling catalyzed by NHC-bis(Ni) complex 33-37.

图21 三齿双核Ni催化的偶联反应38Fig. 21 Cross-copling catalyzed by dinuclear pincer nickel 38.

图22 络合物(iPrNDI)Ni2(C6H6)的合成以及五种氧化还原态39Fig. 22 The synthesis of (iPrNDI)Ni2(C6H6) and five states of oxidation 39.

图23 络合物3n催化的炔烃的硅氢化反应40Fig. 23 Alkyne hydrosilylations catalyzed by complex 3n 40.
同年,Chirik课题组41发现,以氢桥连,双亚胺配体的双核Ni对催化烯烃的硅氢化反应也具有很高的活性(图24)。但是该催化剂在催化烯烃硅氢化反应时,先转化成单核Ni-H物种后,才进入催化循环,完成催化反应。该类型的催化剂对烯烃的加氢反应也具有很高的活性42。
与此同时,Uyeda课题组43发现催化剂3n可以高效、高选择性的促进炔烃的三聚生成1,2,4-三取代的芳环类化合物,产率最高可达到90%,反应选择性也可以达到20 : 1以上(图25)。该反应可以兼容很多类型的官能团,如醛基、羟基、醚、氟以及含氧杂环等。而含氮单核Ni络合物如3o、3p、3q活性会低很多,最多只能得到14%的收率,而且选择性很差,主要得到的是环辛四烯类化合物。

图24 [Ni(iPrDI)(μ-H)]2催化的硅氢化反应41Fig. 24 [Ni(iPrDI)(μ-H)]2 catalytic hydrosilylations 41.
作者通过当量实验对反应机理做了详细的研究(图26),在使用大位阻如——SiMe3、——SiMe2Ph取代的炔烃,没有得到目标产物。分别得到了3r与3s两种络合物。3r是热力学稳定的,70 °C条件下,3r与10当量的三甲基硅乙炔反应24 h后还是稳定存在的。但是当把3r与位阻较小的炔烃反应时,可以以86%的收率得到目标的芳环类产物。3n与三甲基硅乙炔反应生成的单炔配位的络合物3s,在常温下放置24 h后会发生歧化反应生成与3r结构类似的络合物以及络合物3n。由此可见,五元环金属络合物3r是反应的关键中间体。

图25 络合物3n催化炔烃的三聚成环反应43Fig. 25 Alkyne cyclotrimerizations catalyzed by complex 3n 43.

图26 催化剂3n催化炔烃三聚成环反应的机理研究43Fig. 26 The mechanism of complex 3n catalyzed alkyne cyclotrimerizations 43.
作者通过理论计算也说明双核金属催化体系的不同之处:1) 中间体3r与两个金属中心都有作用,这样就限制了金属环的几何形状,不可能有其他的几何构型。2) 由于金属对丁二烯以及炔烃都有作用,有利于[4 + 2]环加成反应。3) 环加成的选择性受到丁二烯π-系统的影响,导致反应选择性大大提高。
随后,该课题组发现这种双核金属Ni催化剂3n对烯烃的还原环丙烷化也具有很高的催化活性(图27,方程式8)44。该反应使用CH2Cl2做亚甲基源,Zn或者ZnEt2作为还原剂,反应条件温和,该催化体系不仅可以兼容苯乙烯及其衍生物,对链状烯烃以及环状烯烃都可以很好的兼容,各种活性较高的官能团如羰基、酰胺、酯等都能在该体系中兼容。该催化剂对催化还原亚乙烯基转移反应也具有很高的活性(图27,方程式9)45。

图27 催化剂3n催化的烯烃的还原环丙烷化反应44,45Fig. 27 Reductive cyclopropanations of alkenes catalyzed by complex 3n 44,45.

图28 络合物3n催化C——C断键反应46,47Fig. 28 Carbon-carbon bond-breaking reaction catalyzed by complex 3n 46,47.

图29 络合物3n催化叠氮化合物的偶联以及与单核镍催化剂活性比较48Fig. 29 The coupling of aryl azides catalyzed by complex 3n and comparison of the catalytic activation with mononuclear Ni catalysts 48.
降冰片二烯的碳——碳σ键较为惰性的,通常不容易发生化学转化。2017年,该课题组46发现3n可以通过对C——C键的氧化加成,实现降冰片二烯C——C的断键(图28)。作者通过当量实验以及理论计算发现,该氧化加成是一个迅速放热的过程,在30 min条件下就能完全转化成3t (图28,方程式10)。当继续把3t与CO作用时,最终以93%的收率得到了稠环[3,3,0]的产物,桥连稠环[2,3,1]的产物完全没有,选择性为100% (图28,方程式11)。最终作者选用Cr(CO)6为羰基源,TMEDA为添加剂,实现了催化转化(图28,方程式12)。随后,该课题组还实现了催化剂3n活化环丙类化合物(图28,方程式13)47。
同年,他们使用该催化剂实现了芳基叠氮化合物的自偶联合成一系列的偶氮化合物(图29)48,该反应体系不需要额外的氧化还原剂,副产物只有N2,可以兼容对氧化还原条件较为敏感的基团。作者使用对三氟甲基苯基叠氮化合物的自偶联为模板反应,比较了单核Ni催化剂与该双核Ni催化剂的活性,研究发现单核Ni催化剂对芳基叠氮化合物的自偶联反应几乎没有什么活性(转化率在23%-40%之间,产率2%-13%),而3n几乎可以达到当量转化,当催化剂当量降低至0.5%时,产率可达到96%。随后,作者进行了机理研究发现,一分子的叠氮化合物首先与催化剂3n形成μ-NAr的中间体,然后该中间体再继续与另一分子的叠氮生成μ-N2Ar2络合物,最后该络合物在溶剂苯的作用下释放出产物和催化剂,完成催化循环。最后的解离过程是该反应的决速步。
5 结论
金属催化与人们的日常生活息息相关,一直是人们关注的热点领域。同核铁系双金属络合物的制备、表征及其性质的研究在近几年得到了迅猛的发展,尽管目前报道的双核体系金属催化剂催化反应的类型还较少,但仍能看出其相较于传统的单核金属催化剂的巨大优势。相比较单核金属催化剂,双核金属催化剂的催化活性以及反应选择性都大大提高,反应条件温和,操作简单,而且还可以实现单核金属催化剂无法完成的催化反应,具有重要的应用前景。但是目前同核铁系络合物在均相催化方面还有一定的局限性,例如催化剂当量较高,机理方面的研究比较滞后,处于研究的初级阶段等。因此同核铁系双金属络合物在均相催化中的应用还需要进一步的开发和应用。
