白羊草叶片和根系干旱胁迫下microRNAs差异表达分析
2019-07-25李春艳董宽虎
李春艳, 董 洁, 钟 华, 董宽虎*
(1.山西农业大学动物科技学院, 山西 太谷 030801;2. 山西省农业科学院畜牧兽医研究所, 山西 太原 030032; 3.北京市科学技术情报研究所, 北京 100044)
众所周知,干旱胁迫常影响植物生长发育,对农作物造成的损失在所有的非生物胁迫中占首位[1]。植物在漫长的进化过程中形成了适应各种胁迫的机制,许多胁迫响应正是通过诱导或抑制miRNAs的表达进而调控相关基因表达来实现的[2]。miRNAs是一类长度约为19~25nt的非编码的单链小分子RNAs,广泛存在于真核生物中。近年来与干旱胁迫相关的miRNA及其靶基因已经在不同植物中得到了验证,如拟南芥[3]、棉花[4]、水稻[5]、苜蓿[6]和小麦[7]等,但这仅限于一些全基因组已知的物种,对于其他植物则研究的很少。
白羊草(B.ischaemum)为禾本科孔颖草属多年生植物,分布广泛,具有固土保水、生命力强,耐旱、高产耐牧、耐践踏、适口性好等优点[8]。多年来对于白羊草的研究主要集中在生产性能[9-10]、营养价值[11]、生理生态[12]及遗传多样性上[13],随着分子生物技术的发展,对于干旱胁迫下白羊草基因的分离和克隆等研究取得一些成绩[14],但对白羊草耐旱的分子机理及相关miRNAs研究甚少,因此,本研究在结合干旱胁迫条件下白羊草叶片和根系转录组数据的基础上,运用高通量Illumina测序技术对干旱胁迫和正常生长条件下白羊草叶片和根系的miRNAs进行测序分析,筛选出响应干旱胁迫的miRNAs,并对其预测的靶基因进行功能分析,为探索白羊草耐旱的分子机制和提供可利用的耐旱基因资源奠定理论基础。
1 材料与方法
1.1 材料的制备
试验材料为采集于太谷县的白羊草种子,盆栽试验在山西农业大学草业科学的日光温室中进行,温室中的平均温度为15~30℃,平均光照强度为26 278 Lx,相对湿度为50%~60%。土壤取自山西农业大学动物科技试验站牧草试验田的耕层土(0~20cm),pH为7.5,田间最大持水量为23.91%。每盆装风干土7 kg,播种量为50粒,正常浇水,苗齐后间苗,每桶留20株长势一致的幼苗。待白羊草幼苗平均生长至20 cm左右时开始干旱处理,处理前一次性浇水,使桶内土壤含水量保持在19.13±3.38%(相当于田间持水量的80%)。对照组持续每天浇水,使土壤含水量保持在田间含水量的80%。实验组不浇水,当叶片开始出现萎蔫土壤的含水量为10.04±2.73%(相当于田间持水量的42%)时,取长势一致的幼苗,分别剪取每株上全部的叶片和根系;对照组取同期没有进行干旱处理的正常生长幼苗的叶片和根系,每个处理重复3次,液氮速冻后—70℃冰箱保存,用于RNA提取。
为了减少随机取样带来的偏差,每次取样从3株独立的个体中取出,提取RNA后等量混合用于后续的建库测序。在这次试验中,我们构建了4个白羊草的小RNA文库,干旱胁迫后的叶片和根系及其对照组的叶片和根系各1个(标记为TL-3、TR-3、CKL-3和CKR-3)。
1.2 样品RNA提取与文库的构建
采用Trizol法提取白羊草干旱胁迫和对照组的叶片和根系样品总RNA,用1%的琼脂糖电泳检测RNA样品是否有降解以及杂质;用2100 RNA Nano 6000 Assay Kit检测RNA样品的完整性和浓度,然后用凯奥K5500分光光度计检测样品纯度。
总RNA样本检测合格后,首先对总RNA进行片段选择,通过胶分离技术收集18~30nt RNA片段;在分离得到的RNA片段的两端分别连接上接头,然后反转录成cDNA,进行PCR扩增建立测序文库;最后,对检验合格的测序文库进行Illumina HiSeq高通量测序,采用SE50测序策略[15]。
1.3 白羊草miRNAs的鉴定、量化和差异表达分析
Illumina测序所得原始序列,通过去除低质量、接头污染和含未知碱基比例大于10%的reads等过程完成数据处理,得到用于后续分析的目标序列。将过滤后的clean reads根据进一步的长度筛选(植物18~30nt),因数据库中没有白羊草基因组和EST序列信息,选择模式植物拟南芥的基因组参考序列,条件设置为允许1个错配。通过基因组比对分析软件SOAP(short oligonucleotide alignment program)将符合条件的clean reads与参考序列进行比对,得到定位信息[16]。匹配较好的reads用于后续分析。之后,将Rfam数据库和GenBank的ncRNA序列比对到基因组[17],得到该物种ncRNA在基因组中的定位信息,将匹配好的Clean reads和ncRNA的定位信息进行匹配,分别注释为rRNA、tRNA、snRNA、snoRNA和其他ncRNA的小RNA。然后将miRbase数据库中的miRNAs序列比对到基因组中,得到该物种目前已知的所有miRNAs在基因组中的定位信息,将剩余未注释匹配好的Clean Reads与miRNAs的定位信息进行完全匹配[18],就可以鉴定已知miRNAs。对于不能与已知注释区域匹配的Clean Reads,采用软件miRDeep2的方法进行新miRNA的预测[19]。针对每个样本,统计与miRNAs匹配的Total Clean Reads的数量,并计算归一化后的表达量RPM(Reads Per Million)值。然后使用DEGseq软件包进行归一化后的差异表达分析,即选取|log2Ratio|≥1且q<0.05的miRNAs作为差异表达miRNAs[20]。
1.4 miRNAs靶基因的预测与功能富集分析
通过psRobot[21]对差异表达显著的miRNAs进行靶基因的预测,并对这些靶基因做GO(Gene Ontology)功能和KEGG(The Kyoto Encyclopedia of Gene and Genome)代谢通路富集分析。GO有三个类别,分别描述基因的分子功能、所处的细胞位置和参与的生物过程。对于差异表达显著的miRNAs所预测的靶基因,可以采用Blast2GO得到每个基因对应的GO条目[22]。将上述靶基因比对到KEGG数据库(http://www.geneome.jp/ kegg)进行通路富集分析,利用KAAS软件找出显著性富集的通路[23]。
1.5 用qRT-PCR法验证miRNAs的表达
为了对miRNAs测序数据进行验证,我们采用qRT-PCR法对随机选取的8个miRNAs表达量进行测定。首先设计特异性的反转录引物RT primer(reverse transcription primer)(表1),然后以建库所剩的总RNA为模板,按照说明书的指导使用M-MuLV Reverse Transcription进行cDNA的合成。然后使用每个miRNA特异性的正向引物和通用的反向引物(表1)进行PCR扩增,qRT-PCR的反应体系为:总体积为20 μl,包括2 μl的cDNA,0.8 μl引物混合物,10μl的1×SYBR Mix。反应程序为两步法:第一步95℃ 3 min,第二步95℃ 5 s,60℃ 20 s,40个循环。每个样品3个重复,内参基因为18 s rRNA[14],基因的相对表达量使用2-ΔΔCt法进行[24]。

表1 qRT-PCR验证实验中选取的miRNAs引物序列Table 1 Primer sequences of selected miRNAs used in qRT-PCR validation experiment
2 结果与分析
2.1 小RNA测序数据分析
本次测序共获得61 609 058 raw reads和43 566 649 clean reads,碱基Q30(碱基错误率小于0.1%)都在95%以上(表2)。我们分析了18~30nt长度分布的小RNA,unique clean reads的长度分布24nt长度具有明显的峰值(图1),这与其他植物的小RNA高通量测序研究结果相一致。
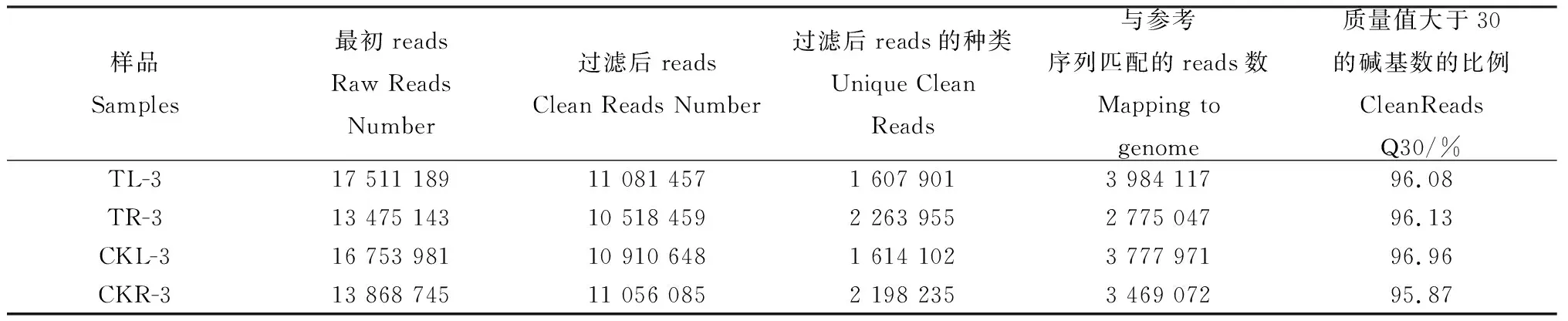
表2 白羊草叶片和根系测序数据统计Table 2 Summary of sequencing in B.ischaemum leaves and roots
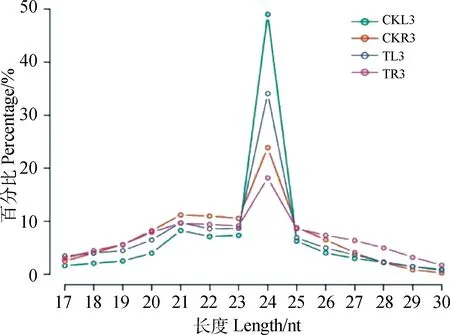
图1 白羊草叶片和根系小RNA过滤后reads种类长度分布Fig.1 Length distribution of unique small RNAs in B.ischaemum leaves and roots
2.2 小RNA分类注释和miRNAs鉴定
Clean Reads中所有比对上小RNA的结果见表2,被注释为miRNAs的平均数量为3 201 445个,达到了总数的7.35%。这表示本实验所构建的小RNA文库已经富集到了白羊草的miRNAs。然而unann的数量平均占到小RNA总数的89.50%,在所有的小RNA种类中最高,这说明本实验所构建的小RNA文库中含有未知的小RNA和潜在的新miRNAs。
总的来说,这次试验鉴定出白羊草中有属于20个miRNAs家族的79个已知miRNAs,在这20个家族中,最大的家族是含有13个成员的ata-miR169家族,其次是ata-miR396含有8个成员,再次是ata-miR156有和ata-miR167均含有7个成员。表达量超过10 000的miRNAs家族有6个,为一些在其它物种中也很保守miRNAs种类。此外,鉴定出92种新miRNA,其中表达量超过500的新miRNAs有10种。
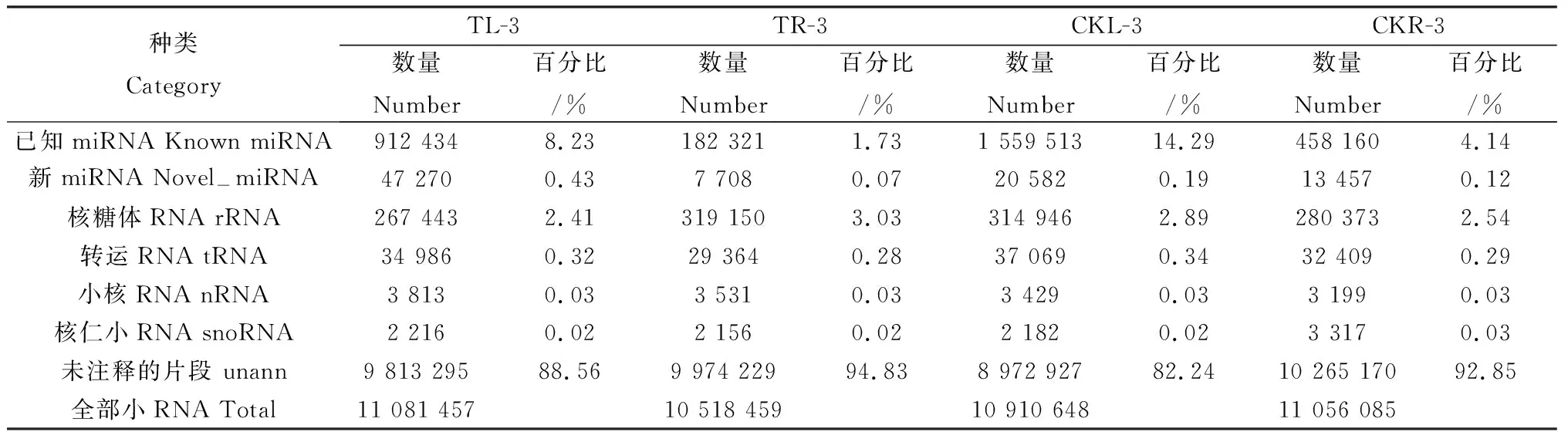
表3 白羊草叶片和根系中小RNA的分类Table 3 Small RNA categorization inB.ischaemum leaves and roots
2.3 miRNAs在干旱胁迫下的差异表达分析
干旱胁迫后叶片和根系miRNAs差异表达数为分别为65和27个(表4),叶片和根系中组织差异的miRNAs分别为55个和17个,共有的差异表达miRNAs为10个(图2)。在这些差异表达的miRNAs中,除了ata-miR156c-3p是在干旱胁迫后的叶片中下调,根系中上调表达外,其余的9个miRNAs在干旱胁迫后的叶片和根系表达模式是一致的,表现为miR164家族和Novel 40均上调,剩余6个miRNAs均下调。

表4 干旱胁迫条件下白羊草叶片和根系差异表达miRNAs的个数Table 4 The number of differentially expressed miRNAs in leaves and roots of B.ischaemum under drought stress
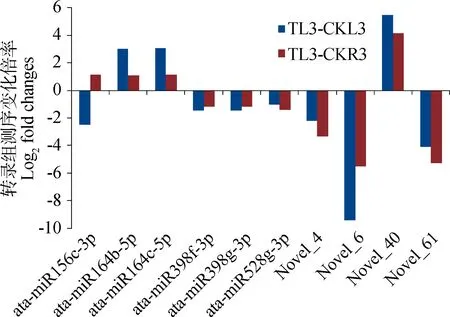
图2 白羊草叶片和根系中共同响应干旱胁迫的miRNAs的表达水平Fig.2 The expression level of drought-responsive miRNAs common in leaves and roots ofB.ischaemum
2.4 差异表达miRNA靶基因预测及GO功能富集分析
miRNAs调控的靶基因鉴定是了解miRNA功能的关键。本次试验干旱胁迫后叶片和根系已知miRNAs预测的靶基因分别为430个和158个,新miRNAs所预测到的靶基因则多达几千个,结合转录组注释结果[25],发现预测的这些靶基因多为转录因子、其他的转录调节物质和一些与胁迫相关的蛋白及与激素信号转导相关的物质。
对这些预测到的靶基因进行GO富集分析(图3),发现无论是叶片还是根系,miRNAs预测到的靶基因在生物学过程中富集基因数最多的是cellular process(GO:0009987),metabolic process(GO:0008152),single-organism process(GO:0044699),分子功能中富集最多的是binding(GO:0005488),catalytic activity(GO:0003824),在细胞组分中富集基因数最多的cell part(GO:0044464),organelle(GO:0043226),membrane(GO:0016020)。其中“cellular process”,“metabolic process”,“cell part”,“binding”和“catalytic”注释到的靶基因数均占所有注释靶基因数的50%以上。

图3 干旱胁迫条件下白羊草差异表达miRNAs预测靶基因的GO功能富集分析Fig.3 Function analysis of target transcripts of differentially expressed miRNAs inB.ischaemum under drought stress.注:横坐标为Ontology分类,纵坐标为注释到该class中的靶基因占所有注释的靶基因的比例Note:The X-axis is the Ontology classification,and the Y-axis is the ratio of the target gene annotated in the class to the target gene annotated in all annotations
2.5 KEGG富集分析
本实验检测出367条通路KEGG通路,其中与干旱胁迫相关且靶基因显著富集的的通路在干旱胁迫后的叶片中有10条,在干旱胁迫后的根系中有1条(表5)。从表5中我们可以看出,Plant hormone signal transduction是干旱胁迫后叶片和根系共有的主要富集代谢通路,而Flavonoid biosynthesis则仅在干旱胁迫后叶片中显著富集且具有最高的Rich factor(指差异表达的基因中位于该pathway条目的基因数目与所有注释基因中位于该pathway条目的基因总数的比值)。
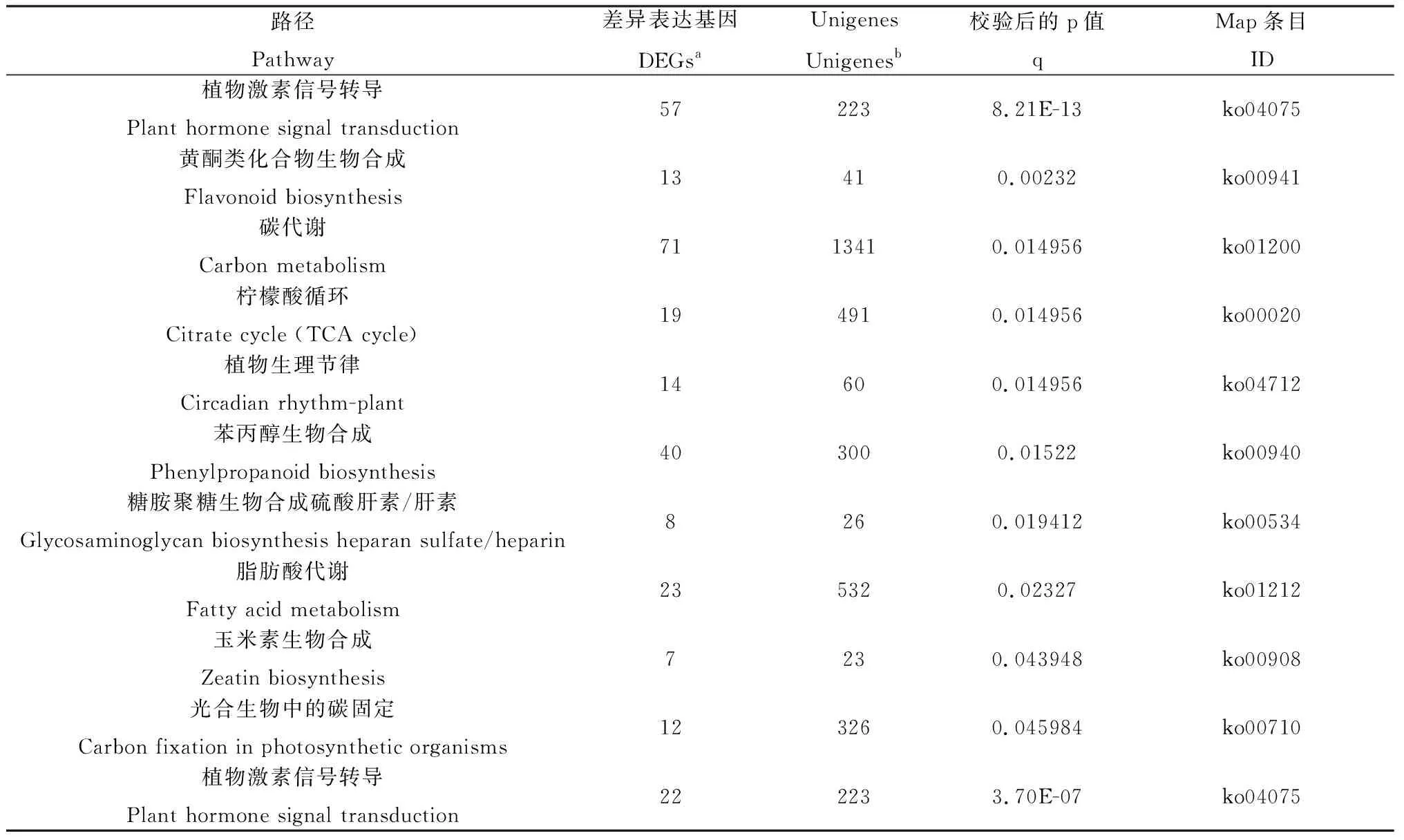
表5 干旱胁迫条件下白羊草叶片和根系(A:叶片;B:根系)代谢通路富集分析Table 5 The enriched pathway in leaves and roots (A:leaves;B:roots) of B.ischaemum under drought stress
注:q≤0.05作为KEGG通路显著富集的标准,q代表调整后的P值,a代表富集到某一特定通路的差异表达基因的数目,b代表富集到该通路上的所有unigene数
Note:The significantly enriched KEGG pathways were determined using q≤0.05,q represents the corrected P-value,a Number of DEGs assigned to certain KEGG pathway,b Number of all reference unigenes assigned to certain KEGG pathway
2.6 miRNAs的qRT-PCR验证
我们对随机选取的8个具有显著差异表达的miRNAs进行定量qRT-PCR验证,从高通量测序miRNAs表达量(Fig.4A)和qRT-PCR(Fig.4B)的结果可以看出,这8个miRNAs在高通测序和qRT-PCR检测中表现出相似的表达模式。
3 讨论
MiRNAs在植物适应逆境胁迫的过程中发挥着重要作用。有一些miRNAs家族在不同植物物种之间是保守的,甚至在一些进化距离较远的苔藓与开花植物之间依然存在保守性[26],但这些保守的miRNA在不同物种和组织中表达是不同的。如干旱胁迫条件下miR398在西红柿[27]和棉花[4]中是下调的,而在小麦[28]和蒺藜苜蓿[6]中则是上调表达。这次研究发现miR398g-3p和miR398f-3p在干旱胁迫后的叶片和根系中均是下调。Kantar在对干旱胁迫后大麦叶片和根系miRNAs的研究中,发现四种miRNAs在脱水条件下呈现出组织特异的调控:miR166在叶中上调根中下调;而miR156a,miR171与miR408在叶中发生了诱导改变表达而在根中则没有变化[29]。这与水稻干旱胁迫后的结果有一些差异,水稻不论是地上部分还是根系在干旱胁迫后miR156和miR171的表达均下调[30]。在本试验中,miR156家族在干旱胁迫后叶片中下调根中上调,miR167,miR172,miR390,miR394,miR408在叶中发生了诱导改变表达在根中没有发生表达变化,而miR166,miR171则是在根中发生了诱导改变表达在叶中没有发生表达变化。这些具有冲突性的结果表明miRNAs的复杂调控并非仅与不同的物种和胁迫条件及组织差异性有关,也与物种在长期进化适应过程中所形成的同一家族中不同miRNAs成员的差异有关。

图4 白羊草叶片和根系响应干旱胁迫的miRNA定量PCR验证Fig.4 qRT-PCR validation of drought-responsive miRNAs in leaves and roots of B. ischaemum
研究表明,保守的miRNAs在大量物种中都有保守的靶向基因[31],这表明miRNAs与其所调控靶基因之间的关系在植物长期进化的过程中趋于稳定[32],如miR156的靶基因SPL (squamosa promoter-binding-like protein)在植物发育过程、合成代谢及非生物胁迫中起到了重要的作用,成为植物生长与发育的调控枢纽[33]。我们这次对白羊草miR156靶基因的预测表明,SPL是miR156所预测的主要靶基因。但除SPL外,我们所预测的miR156还有一些新的靶基因如脂质转运蛋白、质膜ATPase等。预测的miR164的靶基因除了在大多数植物中已经验证了的NAC转录因子,还有丝氨酸-苏氨酸蛋白激酶和假想蛋白等。虽然这些新预测的靶基因还有待进一步的验证,但靶基因预测的结果给我们一个暗示,一个miRNA家族中不同的成员可能能够调控各自不同但是较为相似的靶基因,从而参与多个生理生化途径的调控。
虽然一些新预测的miRNAs的成熟序列在miRBase中并未找到对应的序列,但通过对其预测的靶基因进行GO功能聚类分析和KEGG代谢途径分析得知,这些新miRNAs主要在植物生长、发育、胁迫响应和激素信号转导中发挥作用。如miRNA_39被预测可调控植物激素信号转导中ABA信号通路,miRNA_36被预测与白羊草叶片响应干旱胁迫的黄酮类化合物代谢途径密切相关。这说明这些新预测的miRNAs对白羊草适应干旱胁迫有着重要的作用,研究和探索这些新预测miRNAs的表达模式和功能对进一步阐明白羊草适应干旱的分子机制具有重要意义。
4 结论
这次通过对干旱胁迫条件下白羊草叶片和根系miRNAs表达特性的研究发现,白羊草响应干旱胁迫的miRNAs除了一些在其他植物中已经被证实与干旱胁迫相关的miRNAs外,还发现一些与干旱胁迫相关的新miRNAs,通过对miRNAs靶基因生物学功能和代谢途径的富集分析发现,植物的次生代谢产物尤其是黄酮类化合物合成和植物信号转导途径是白羊草响应干旱胁迫富集的主要通路。
