烟草NtTkr尾部T1084缺失和替换突变原核表达载体的构建及诱导表达
2019-07-05童文艳胡孟可徐林娜乔慧聪
童文艳,胡孟可,徐林娜,乔慧聪,李 芬
(河南师范大学 生命科学学院,河南 新乡 453007)
NtTkr是新发现的烟草驱动蛋白超家族新成员,属于组织特异性基因,仅在分裂旺盛组织优势表达,参与细胞周期调控,与胚、种子大小和萌发有关[1-3]。驱动蛋白种类繁多[4-6],功能多样,不仅与细胞运动、细胞分裂、形态建成、胞内物质定向运输等众多生物过程相关,对大脑发育、记忆提升、神经元活性也很重要[7-8],是维持细胞活性的关键蛋白质,这些功能均与其尾部卷曲螺旋结构(Coiled-coil,CC)密切相关。
NtTkr尾部共有3个卷曲螺旋结构(CC1、CC2、CC3),其中CC1、CC2 参与茎秆结构的形成,CC3(1049-1143 Aa)负责与底物的结合[2]。酵母双杂交分析表明,CC3的T1084突变或缺失会影响NtTkr与靶蛋白的结合强度。为确定T1084缺失或替换突变对NtTkr与候选蛋白体外互作的影响,采用重叠延伸PCR在烟草NtTkr尾部引入T1084缺失和替换点突变,并将突变的NtTkr尾部克隆入pMXB10质粒,构建重组载体pMXB10-T1084d和pMXB10-T1084A,最后将其转入BL21(DE3)中,IPTG成功诱导NtTkr-T1084A-1320和NtTkr-T1084d-1317表达,为确定NtTkr与候选蛋白之间的体外互作及关键结构域奠定基础。
1 材料和方法
1.1 材料、试剂及药品
材料:菌株AH109、Y187,质粒pBI121-NtTkr、pUC19、pMXB10,大肠杆菌DH5α、BL21、DE3等。
主要试剂及药品:SD及缺陷培养基、2×SDS上样缓冲液、X-Gal(Clontech公司,美国);KOD-plus高保真酶(TIANGEN,北京);dNTPs(2 mmol/L each)、10×KOD-plus Buffer、25 mmol/L MgSO4(ToYoBo);DNA Marker λ-Eco T14(TIANGEN,北京);6×Loading Buffer、限制性内切酶、T4-DNA连接酶(TaKaRa,日本);质粒提取试剂盒(TIANGEN,北京);DNA凝胶回收试剂盒(TaKaRa,日本);鲑鱼精DNA(上海浩然生物技术有限公司)。
1.2 方法
1.2.1 目的基因片段的扩增与检测 缺失和替换突变采用3对引物的3轮PCR来完成,引物由TaKaRa生物公司合成。目的基因扩增引物序列为:P1:5′-CATATGGATGGTCCCTGTGGAAGG-3′,P2:5′-GCGGCCGCTATGCGTTCCTGGTAAATGGC-3′,在P1和P2两端分别引入NotⅠ和NdeⅠ酶切位点;替换突变(T1084A)位点的中间互补引物序列为P3′:5′-CTCAACTTAAAGACGCTGCTGAAGCTGTTC-3′,P4:5′-GAACAGCTTCAGCAGCGTCTTTAAGTTGAG-3′;缺失突变(T1084d)位点的中间互补引物序列为P5:5′-CTCAACTTAAAGACGCTGAAGCTGTTC-3′,P6:5′-GAACAGCTTCAGCGTCTTTAAGTTGAG-3′。以质粒 pBI121-NtTkr为模板,分别以P1、P3,P2、P4及P1、P5,P2、P6为引物进行PCR扩增,得到的扩增产物经凝胶回收后等摩尔混合,取适量作为模板,以P1、P2为引物扩增完整尾部片段。
1.2.2 表达载体pMXB10-T1084A和pMXB10-T1084d的构建和鉴定 凝胶电泳回收SmaⅠ线性化的pUC19和PCR产物,取适量回收产物在T4DNA连接酶作用下16 ℃连接16 h,连接产物转化大肠杆菌感受态DH5α细胞,涂AMP+和X-gal选择平板,经NotⅠ-NdeⅠ双酶切鉴定获得正确重组子,并进行基因组测序。将pMXB10与测序正确的pUC19-T1084d、pUC19-T1084A分别进行NotⅠ-NdeⅠ双酶切,回收6.68 kb的pMXB10载体片段和约1.32 kb的突变目的片段,在T4DNA连接酶作用下16 ℃连接16 h后转化大肠杆菌超级感受态细胞BL21,NotⅠ-NdeⅠ双酶切鉴定。
1.2.3 目的蛋白的诱导表达SDS-PAGE检测 将鉴定后的pMXB10-T1084A和pMXB10-T1084d载体转化入BL21(DE3)超级感受态细胞,在50 mg/mL氨苄青霉素LB平板上挑取单克隆,37 ℃、220 r/min 过夜培养,次日以1∶100稀释菌液,培养4 h左右至OD600为0.6~0.8,分别经0,0.04,0.06,0.10,0.20 mmol/L和0,0.05,0.10,0.50 mmol/L的IPTG于37 ℃诱导4 h后,取菌液2 mL,于4 ℃、1 000 r/min离心1 min,弃上清。在得到的菌体沉淀中加入还原型2×SDS上样缓冲液和去离子水各100 μL,混匀后煮沸10 min,取15 μL样品进行SDS-PAGE分析。
2 结果与分析
2.1 T1084d和T1084A突变尾部的获得及克隆入pUC19的鉴定结果
以pBI121-NtTkr为模板PCR扩增1 kb和350 bp的T1084上、下游片段(图1-A)及以两片段为模板进行PCR扩增得到的T1084d、T1084A片段(图1-B),结果与预期长度相符。将T1084d、T1084A克隆入SmaⅠ线性化的pUC19,经蓝白斑试验筛选获得重组子后进行SmaⅠ-BamHⅠ双酶切鉴定,得到约1.32 kb的插入片段(图1-C),与突变尾部1 317,1 320 bp编码区符合。测序结果显示,编码第1 084 位苏氨酸已经缺失或为丙氨酸替代。
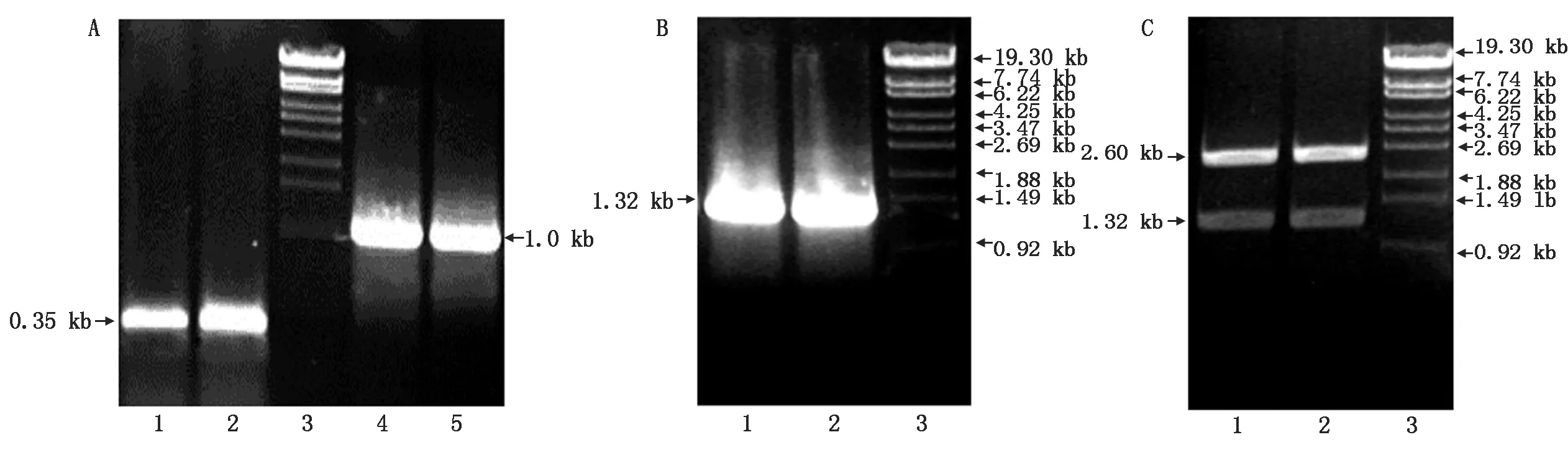
A.缺失和替换突变短片段PCR产物:1,2.克隆得到的T1084d和T1084A 的350 bp短片段;3.DNA标准分子质量λEcoT14;4,5.克隆T1084d和T1084A的1 kb长片段。B.缺失和替换突变完整片段PCR产物:1,2.1.32 kb的T1084d和T1084A片段;3.DNA标准分子质量λEcoT14。C.缺失和替换突变尾部克隆入pUC19后的酶切鉴定:1,2.NotⅠ-NdeⅠ双酶切后的pUC19载体和约1.32 kb的T1084d和T1084A片段;3.DNA标准分子质量λEcoT14。
A.Short fragment PCR products of deletion and replacement mutant:1,2.The cloned 350 bp short fragment of T1084dand T1084A; 3.DNA standard molecular mass λEcoT14; 4,5.The cloned 1 kb long segments of T1084dand T1084A.B.Complete fragment PCR products of deletion and replacement mutant:1,2.1.32 kb fragment of T1084dand T1084A; 3.DNA standard molecular mass λEcoT14.C.Restriction analysis of the deleted and replaced mutant tail cloned into pUC19:1,2.pUC19 vector after theNotⅠ-NdeⅠ double enzymed and about 1.32 kb T1084dand T1084A fragments; 3.DNA standard molecular mass λEcoT14.
图1 T1084d和T1084A的获得及克隆入pUC19后的鉴定结果
Fig.1 Acquisitin of T1084dand T1084A and identification results after cloning into pUC19
2.2 pMXB10-T1084A和pMXB10-T1084d的构建和鉴定结果
将测序正确的重组载体pUC19-T1084d、pUC19-T1084A均与质粒pMXB10进行NotⅠ-NdeⅠ双酶切,分别回收约1.32 kb的目的片段和6.68 kb的载体片段并进行连接。连接产物转化大肠杆菌感受态DH5α并筛选得到重组子,再进行NotⅠ-NdeⅠ双酶切鉴定,电泳结果显示得到了预期片段(图2),表明获得了所需的T1084d、T1084A与pMXB10诱饵表达质粒。

A.原核表达质粒的酶切鉴定:1. DNA标准分子质量λEcoT14;2,3.NotⅠ-NdeⅠ双酶切后6.68 kb的pMXB10载体和1.32 kb的T1084d和T1084A片段。B.pMXB10-NtTkr-T1084d-1317/1084A-1320图谱,显示大小约1.32 kb的突变尾部插入pMXB10的酶切位点。
A.Enzymed digestion and identification of prokaryotic expression plasmid:1.DNA standard molecular mass λEcoT14 ; 2,3. 6.68 kb pMXB10 vector ofNotⅠ-NdeⅠ doubled enzyme digestion and 1.32 kb T1084dand T1084A fragment.B.pMXB10-NtTkr-T1084d-1317/1084A-1320 map,showing that the mutant tail of about 1.32 kb was inserted into the enzyme digestion site of pMXB10.
图2 原核表达质粒pMXB10-T1084d、pMXB10-T1084A的酶切鉴定结果
Fig.2 Enzyme digestion identification of prokaryotic expression plasmid pMXB10-T1084dand pMXB10-T1084A
2.3 NtTkr-T1084A-1320和NtTkr-T1084d-1317的诱导表达
将重组载体pMXB10-NtTkr-T1084A和pMXB10-NtTkr-T1084d分别转化大肠杆菌BL21(DE3)感受态,37 ℃振荡培养至OD600为0.6~0.8,设置IPTG浓度梯度、220 r/min、37 ℃诱导4 h后收集菌体,稀释煮沸后用12% SDS-PAGE检测蛋白质表达情况(图3)。由图3-A可知,IPTG浓度大于0.06 mmol/L时,诱导表达的蛋白质表达量差异不大,均可高效表达约77 ku的NtTkr-T1084A-1320蛋白质;由图3-B可知,IPTG浓度大于0.05 mmol/L,诱导后目的蛋白的表达量几乎相当,均可高效表达约76.2 ku的NtTkr-T1084d-1317蛋白质。
3 结论与讨论
NtTkr是一个在幼叶和发育各时期胚等分裂旺盛组织中优势表达的驱动家族的新成员[1-3]。具有组织特异性NtTkr可能通过与细胞内多种蛋白质相互作用参与基质中合成蛋白的定向运输、加工修饰、特定蛋白质的降解以及重金属毒害的解除等诸多生物学过程,而这些功能都与分子内或分子间的卷曲螺旋结构有着直接关系[9-10]。已知NtTkr有3个卷曲螺旋结构,而且位于尾部的CC3(1 049-1 143 Aa)很有可能在与靶蛋白的相互作用中发挥功能。研究已经证实,CC3结构域中1 084位苏氨酸的缺失和替换,转入洋葱表皮细胞其定位会发生变化,酵母双杂交也证实上述突变会改变与候选蛋白互作的强度[11-12],但目前对其与底物互作的关键结构域还缺乏认识,进一步运用Pull Down等手段验证其与候选蛋白的互作非常必要[13-15],而pMXB10-T1084d和pMXB10-T1084A成功构建和诱导表达为运用Pull Down确定NtTkr与候选蛋白之间的体外互作以及关键结构域奠定了基础。
IMPACT系统是一种应用广泛的新型蛋白质融合表达及纯化系统,能应用到蛋白质工程的许多重要领域,可高效诱导且方便后期纯化获得较高纯度的目的蛋白[16-18]。故本研究按照经济实用的原则,采用了该系统pMXB10质粒,通过重叠延伸PCR在烟草NtTkr尾部引入T1084缺失(T1084d)和替换(T1084A)点突变,并将其分别克隆入pMXB10质粒,测序及酶切验证表明,成功构建了将T1084A-1320和T1084d-1317克隆入pMXB10的融合表达载体。

A.NtTkr-T1084A-1320的诱导表达:1.蛋白质分子质量Marker;2-6.浓度依次为0,0.04,0.06,0.10,0.20 mmol/L的IPTG诱导后NtTkr-T1084A-1320蛋白的表达。B.NtTkr-T1084d-1317的诱导表达:1-4.浓度依次为0.50,0.10,0.05,0 mmol/L的IPTG诱导后NtTkr-T1084d-1317蛋白的表达;5.蛋白质分子质量Marker。
A.Induced expression of NtTkr-T1084A-1320:1.Protein molecular mass Marker; 2-6.Expression of NtTkr-T1084A-1320 protein induced by IPTG at concentrations of 0,0.04,0.06,0.10,0.20 mmol/L.B.Induced expression of NtTkr-T1084d-1317:1-4.Protein expression of NtTkr-T1084d-1317 induced by IPTG at concentrations of 0.50,0.10,0.05,0 mmol/L; 5.Protein molecular mass Marker.
图3 NtTkr-T1084A和NtTkr-T1084d的诱导表达结果
Fig.3 Induced expression of NtTkr-T1084A and NtTkr-T1084d
成功构建重组原核表达载体后,将其转化大肠杆菌BL21(DE3)超级感受态细胞并进行IPTG诱导表达。目的蛋白的可溶性、稳定性和表达量因蛋白质而异,因此诱导表达的时间和温度可根据实际情况进行调整。为了使蛋白质达到比较大的表达量,研究中尝试摸索了最佳的培养条件。如,对于T1084A的诱导,设置了5个不同浓度梯度的IPTG,蛋白质电泳检测结果发现,IPTG浓度为0.06 mmol/L时的表达量明显大于0.04 mmol/L时,但却与0.1,0.2 mmol/L时相差不大,表明IPTG浓度为0.06 mmol/L较为适宜。在诱导温度的选择上,在相同条件下,尝试了37 ℃诱导4 h、30 ℃诱导6 h、22 ℃诱导过夜。结果显示,37 ℃诱导4 h蛋白质的表达量相对较大。经过上述摸索,选择的最优诱导表达条件为:220 r/min、0.06 mmol/L IPTG于37 ℃诱导4 h,目的蛋白NtTkr-T1084A-1320得到了高效表达。运用类似的方法发现,220 r/min、0.05 mmol/L IPTG于37 ℃诱导4 h,可以实现NtTkr-T1084d-1317 的高效表达。
综上,成功构建了NtTkr-T1084A-1320和NtTkr-T1084d-1317克隆入pMXB10的融合表达载体并诱导其高效表达,上述两突变蛋白质的成功表达为下一步的蛋白质纯化、Pull Down确定NtTkr与候选蛋白之间的体外互作进而确定NtTkr与蛋白质互作的关键结构域奠定了基础。
