Expanding etiology of progressive familial intrahepatic cholestasis
2019-06-20SarahAFHenkelJudySquiresMaryAyersArmandoGanozaPatrickMckiernanJamesSquires
Sarah AF Henkel, Judy H Squires, Mary Ayers, Armando Ganoza, Patrick Mckiernan, James E Squires
Sarah AF Henkel, Division of Gastroenterology, Hepatology, and Nutrition, Emory School of Medicine, Atlanta, GA 30322, United States
Judy H Squires, Department of Radiology, UPMC Children’s Hospital of Pittsburgh,Pittsburgh, PA 15224, United States
Mary Ayers, Patrick Mckiernan, James E Squires, Division of Gastroenterology, Hepatology,and Nutrition, UPMC Children’s Hospital of Pittsburgh, Pittsburgh, PA 15224, United States
Armando Ganoza, Division of Pediatric Transplantation, Department of Surgery, UPMC Children’s Hospital of Pittsburgh, Pittsburgh, PA 15224, United States
Abstract
Key words:Cholestasis; Progressive familial intrahepatic cholestasis; Benign recurrent intrahepatic cholestasis; Intrahepatic cholestasis of pregnancy; Drug induced cholestasis;Bile acids; Bile transport
INTRODUCTION
Progressive familial intrahepatic cholestasis (PFIC) refers to a heterogeneous group of autosomal recessive disorders that are linked by the inability to appropriately form and excrete bile from hepatocytes, resulting in a hepatocellular form of cholestasis.While the diagnosis of such disorders had historically been based on pattern recognition of unremitting cholestasis without other identified molecular or anatomic cause, recent scientific advancements have uncovered multiple specific responsible proteins. The variety of identified defects has resulted in an ever-broadening phenotypic spectrum, ranging from traditional benign recurrent jaundice to progressive cholestasis and end-stage liver disease.
Bile is a unique aqueous secretion of the liver that is formed by the hepatocyte and modified downstream by absorptive and secretory properties of the bile duct epithelium. It is a combination of lipids (mainly phosphatidylcholine), bile acids,cholesterol, bilirubin, and other substances that serve to move toxins and waste metabolites out of the liver and into the gut for excretion[1]. Micellarized bile is then reabsorbed in the enterohepatic circulation in the distal small bowelviathe apical sodium dependent bile transporter (ASBT;SLC10A)[2]. Bile salts are synthesized in hepatocytes and transported across the canalicular membraneviathe bile salt export pump (BSEP); the expression and trafficking of which is regulated by the farnesoid X receptor (FXR) and dependent upon of MYO5B respectively[3,4]. The stability of the canalicular membrane, in which the BSEP transporter lies, is dependent on the FIC1 ATPase that regulates the phospholipid balance and the ABC translocase MDR3 which moves phosphatidylcholine across the canalicular membrane to inactivate bile acids. The integrity of the system is in part dependent upon hepatocyte connections,such as the TJP2-anchored tight junctions, which protect hepatocytes from bile salt reflux and subsequent damage[4](Figure 1). Defects in these bile acid transport processes result in the accumulation of bile salts in the hepatic parenchyma, which aretoxic due to their detergent nature, and the phenotypic manifestations collectively known as PFIC.
MATERIALS AND METHODS
This systematic review was conducted according to the PRISMA guidelines. We searched Medline/PubMed in February-March 2019 for established cases of PFIC as well as reports of defects in PFIC-related genes contributing to morbidity in adult populations. English language only articles that were fully accessible were included in the review. Data was manually extracted on disease characteristics in established PFIC patients. Associated phenotypes with other diseases relating to specific genetic defects were also collected. Treatment strategies were summarized. Data was collated and presented in text, figure, and table format.
Statistical analysis
Descriptive statistics were utilized to present the data. The statistical methods of this study were reviewed by Suraj Nepal, lead data analyst from the UPMC Children’s Hospital of Pittsburgh department of surgery.
RESULTS
A summary of currently understood protein mechanisms, whose functions are critical to bile acid homeostasis, and whose dysfunction results in a phenotype of PFIC is presented in Figure 1. A gene-specific search identified 52ATP8B1, 158ABCB11, 250ABCB4, 56TJP2, 48MYO5B, and 363NR1H4articles. Manual review to identify association with liver disease in humans revealed reports summarized in the current manuscript. The three “Historical” PFIC diseases, the expanded phenotypes, and emerging data on contributing morbidity in non-pediatric populations relating to defects in PFIC-related genes are summarized.
Historical PFIC
ATP8B1(FIC1, PFIC1, Byler’s disease):The first reported PFIC, progressive familial intrahepatic cholestasis type 1, also called Byler’s disease, was described 1969 in seven Amish children (from the original Byler kindred in Western Pennsylvania) as a progressive cholestatic disease with associated extrahepatic symptoms[5]. The causativeATP8B1gene and corresponding FIC1 protein was identified by Bullet al[6]in 1998 by analyzing the genetics of patients from the initial Amish cohort as well as patients from Northern Europe with benign recurrent intrahepatic cholestasis type 1(BRIC1). Definitive FIC1 function remains ambiguous. Current understanding of its action as an aminophospholipid translocase which transports phospholipids from outside to inside the canalicular membrane is based on studies in Atp8b1-deficient mice[7]. Additional modifiers of disease phenotype, such as mutation-specific effects on FIC1 trafficking from the endoplasmic reticulum to the canalicular membrane,have been proposed[8]. Ultimately, without appropriate concentrations of intracellular phospholipids, bile acids accumulate intracellularly and are cytotoxic to the hepatocyte due to their detergent nature[9].
Deficient or defective FIC1 results in a low gamma glutamyl (GGT) cholestasis that often presents in the neonatal period, though milder forms with transient jaundice may present later in life[1,9,10]. Affected individuals have hyperbilirubinemia, mildly elevated transaminases, and elevated serum bile acids. Infants often present jaundiced, with pruritis and hepatosplenomegaly developing over the first months of life. Severe disease manifests with persistent, progressive cholestasis and the development of portal hypertension often in early childhood. Extrahepatic disease is also notable due to the broad distribution of FIC1, which can clinically distinguish FIC1 deficiency from other forms of intrahepatic cholestasis. Affected children frequently exhibit profound diarrhea, poor growth, short stature, pancreatic insufficiency, elevated sweat chloride, and sensorineural deafness[4,10]. Histopathology demonstrates canalicular cholestasis with biliary plugs, giant cell transformation,ductular paucity, and lobular disarray[11]. Visualized bile is termed as “bland”granular (Byler’s) bile[9,12].

Figure 1 Molecular mechanisms of cholestasis at the apical hepatocellular membrane.
Treatment for FIC1 deficiency, as with all PFIC diseases, is challenging with no definitive medical therapies available. Supportive measures are focused on improving nutritional deficiencies and managing complications of end stage liver disease.Patients should be treated with caloric, fat, and vitamin supplementation, with the majority of fat being medium chain triglycerides[9]. Ursodeoxycholic (UDCA), a hydrophilic bile acid which replaces hydrophobic bile salts and may also induce BSEP and MDR3 expression, can improve pruritis and biochemical markers of cholestasis[9].Other antipruritic agents (Table 1) such as rifampin and cholestyramine may also be utilized but are often less helpful in FIC1 deficiency[9,10]. Certain CFTR folding correctors have been shown to improve defective trafficking of FIC1 in cell culture[13];however, studies in human subjects are lacking.
1996年,适逢团场机车改制,在很多机车驾驶员犹豫时,孙改会不管不顾,硬是靠东挪西凑的钱,在连队带头把他那辆开惯了的铁牛55拖拉机买了下来。同时,他和妻子承包的棉花地也增加到了100余亩。尽管孙改会领着妻子每天忙得跟个陀螺似地,但一年年不断递增的经济收入让他把想要创业、创新的视野和心胸放得更长、更远。
When medical therapy is insufficient, surgical intervention may be considered with the goal of bypassing the enterohepatic circulation and/or decreasing reabsorption of bile salts (Figure 2). Procedures including partial external biliary diversion (PEBD),partial internal biliary diversion, and ileal exclusion have generally, though not uniformly, resulted in sustained clinical improvement in PFIC patients[14-16]. A large surgical experience has been described in FIC1 deficiency[14-16].
Early procedures including PEBD were first reported more than 20 years ago[17].PEBD utilizes an external stomal conduit (generally a cholecystojejunal cutaneous stoma) to enable partial, unregulated external bile flow, resulting in decreased bile acids in the enterohepatic circulation and reports of improved pruritis, growth, and possibly hepatic fibrosis[14,17]. Remarkably, PEBD has been recognized to provide an alternative to transplant, with many patients surviving with their native liver.However, complications can occur including recurrent episodes of pruritis, possible need for biliary diversion revision, continued need for aggressive vitamin supplementation, or progressive disease necessitating liver transplant[14,18,19].
An alternative to PEBD is the ileal bypass (IB, or ileal exclusion)[20]. This technique bypasses the distal 15% of the ileum to avoid the major site of bile acid reabsorption and is particularly useful in patients without an intact gall bladder[20]. Unfortunately,severe malabsorption can occur and refractory disease has been reported[18,21,22].
More recently, partial internal biliary diversions (PIBD) has been described. The procedure may involve the creation of a neo-conduit between the gall bladder and the colon to prevent reabsorption of bile acids in the terminal ileum. This procedure may utilize a cholecystojejunocolonic, cholecystoileocolonic, cholecystocolostomy, or cholecystoappendicocolonic anastomosis technique[22,23]. Reports in the literaturesuggest patients experience not only improvement of intractable pruritis and sleeping difficulties, but also significant biochemical decrease in both bilirubin and plasma bile acids[21-23]. Side effects described are most notable for diarrhea, which improved with cholestyramine[21,23]. Notably, no single procedure has demonstrated definitive superiority with center-experience likely driving center-specific approaches. Newer therapeutics including inhibitors of the ileal apical-sodium dependent bile acid transporter (ASBT) which effectively act as a ‘chemical’ biliary diversion are currently under investigation (NCT03566238)[24].

Table 1 Medical management of pruritus in children
Liver transplant is indicated in those with a refractory course and in those who develop end stage disease. While hepatocellular carcinoma as an indication for transplant has been reported in other PFIC diseases, FIC1 deficiency is not known to associate with tumor development. However, mutations inATP8B1have been found while sequencing hepatocellular carcinoma in patients without cholestatic disease[25].Importantly, patients should be counseled that the diarrhea associated with FIC1 deficiency may persist, or even worsen, following transplant. This phenomenon has been reported concomitant with the development of both allograft steatosis and fibrosis, which can progress requiring re-transplantation[9,26]. In order to prevent damaging steatosis in the graft, ileal diversion at the time of transplant has sometimes been utilized[27].
Notably, the recognition of variable disease courses and responses to therapy in individuals with identicalATP8B1mutations would suggest the presence of disease modifiers[10,14]. While the majority of FIC1 deficiency presents in childhood, mutations in theATP8B1gene may also lead to more mild manifestations of disease including BRIC1 and intrahepatic cholestasis of pregnancy type 1 (ICP1)[28-30]. Dozens of mutations have been described, with missense mutations being more common in BRIC1 patients and nonsense or large deletions more common in severe FIC1 disease[31].
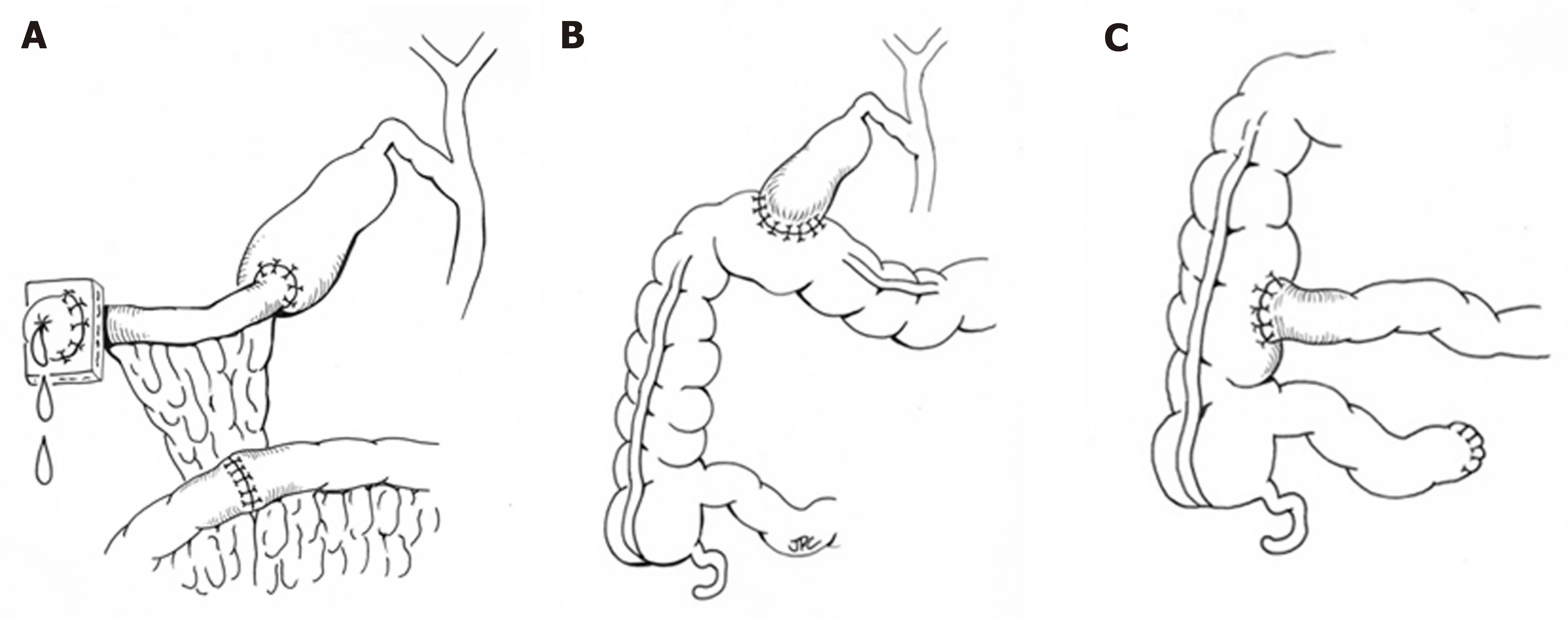
Figure 2 Representative surgical interventions for progressive familial cholestasis.
ABCB11(BSEP, PFIC2):Historical PFIC2 results from defects or deficiency in the BSEP encoded byABCB11. The location of the defect was initially mapped to chromosome 2q24 to be positional match to BSEP, which had been cloned previously in the mouse genome and was shown soon after to export bile acids[32,33]. This defect results in a severe hepatobiliary phenotype due to impairment of bile salt handling and subsequent damage to hepatocytes[9]. As of this writing, more than 200 causative mutations have been identified[34]. Affected infants initially present jaundiced, with pruritis developing around 4-5 mo of age and often progressing to the development of portal hypertension within the first year of life[9,35,36]. Scleral icterus, hepatomegaly,excoriation of skin, and poor growth due to fat malabsorption and fat-soluble vitamin deficiency may also be apparent due to cholestasis, though extrahepatic symptoms are less significant than in FIC1 deficiency[4,9]. Laboratory findings demonstrate a low GGT cholestasis with transaminases typically more than twice the upper limit of normal[36]. Similar to FIC1 deficiency, treatment is primarily supportive and focuses on nutritional supplementation and antipruritic agents. Zebrafish models of BSEP deficiency suggest a potential role for therapies aimed at promoting alternative transporters to excrete bile[37]while reports in human subjects using cell surface BSEP-enhancer molecules (i.e., 4-phenylbutyrate) alone[38]or as part of a cocktail of medications[39]have shown promise. Both approaches require more complete investigation, which may be facilitated through new disease models using patientspecific induced pluripotent stem cell-derived hepatocyte like cells[40]. Surgical interruption of the enterohepatic circulation may improve pruritis but may not change the course of disease[16]. Notably, the response to diversion has been shown to be dependent on the gene defect, with those who retain some residual protein function having better outcomes than those with mutations resulting in severely dysfunctional or absent protein[41,42]. Pathology typically demonstrates canalicular cholestasis, hepatocellular disarray, and lobular and portal fibrosis[9]. Importantly,there is up to 15% rate of malignancy (hepatocellular carcinoma and cholangiocarcinoma) that has been described in children as young as 13 mo[43]. Therefore,patients with PFIC2 should be screened for malignancy with an alpha-fetoprotein(AFP) level and abdominal ultrasound every 6-12 mo[9]. Liver transplant has been successfully used to treat severe BSEP disease and in those who develop tumor. While organ replacement has historically been considered a ‘’cure’’, patients can develop allo-reactive antibodies specific to the extracellular loop of the BSEP protein resulting in an immune mediated recurrence of their BSEP disease in the allograft[44-46].Monitoring for disease recurrence is critical as most disease will respond to increased immunosuppression. However, with refractory disease recurrence, more intensive management such as B-cell depleting antibody therapy[47], allogenic hematopoietic stem cell transplant[48], and repeat solid organ transplant[49]may be required.
Similar toATP8B1, a phenotypic continuum has been recognized withABCB11mutations. Transient neonatal cholestasis, benign recurrent intrahepatic cholestasis type 2 (BRIC2), intrahepatic cholestasis of pregnancy type 2 (ICP2), and drug induced cholestasis have all been associated with abnormalities in BSEP[50,51]. Two mutations have been found that prognosticate a modified disease course of BSEP disease:D482G leads to a more slowly progressive disease with the development of cirrhosis at a later age, and E297G results in PFIC2 or BRIC2 that may be more responsive to medical therapy[4,9]. Drug induced cholestasis is often associated with the V444A mutation,which leads to decreased BSEP expression, and specifically contraceptive induced cholestasis has been associated with the 1331T>C polymorphism[52,53].
ABCB4(MDR3, PFIC3):Also described as a cholangiopathy, PFIC3 is secondary to defects in the multidrug resistance class 3 (MDR3) glycoprotein, encoded byABCB4[54].As a phospholipid translocator, MDR3 facilitates the incorporation of phosphatidylcholine into bile. Without phosphatidylcholine to neutralize bile acids,the imbalance of free bile acids damages cholangiocytes, and cholesterol crystallizes into liver-damaging stones[9]. As with other PFIC diseases, there is a spectrum of disease that can be explained by the extent to which MDR3 is impaired by a particular genetic mutation[55,56]. Those with a heterozygous mutation typically have a milddisease course, including forms of transient neonatal cholestasis[55,57]. Of the described defects in MDR3, the majority are missense mutations that result in defective processing or intracellular transport; while the minority have completely absent MDR3 expression secondary to early truncation or destruction of the protein[9,55,57,58].While presentation in the first months of life are reported, MDR3 deficiency more often presents in late adolescence or even adulthood[59]. The phenotype of adults withABCB4mutations can be varied, ranging from slowly progressive disease,cholelithiasis, ICP, drug induced cholestasis, and benign recurrent intrahepatic cholestasis[58]. In children and adolescents, symptoms are typically few, and the first may be variceal bleeding secondary to portal hypertension[9]. A retrospective review of 38 patients found that those diagnosed in childhood presented with pruritis around 1 year of age and most had hepatosplenomegaly, portal hypertension, and jaundice at the time of presentation[58]. Pediatric disease has also been associated with growth restriction, reduced bone density, and learning disabilities[58]. GGT is typically elevated at presentation, with relatively milder elevation of transaminases and bilirubin[41,59]. Medical treatment should be initiated early in the disease course. Care is supportive including nutrition supplementation and antipruritic agents, though it is not clear if these therapies alter the disease course[4,55]. In vitro studies have suggested that disease-associated mutations resulting in impaired ABCB4 trafficking may be functionally rescued by chemical chaperones[56]. Temporizing surgical interventions as described above are rarely successful due to the severity of disease when diagnosed and liver transplant remains the only definitive therapy[4,59]. Histology typically demonstrates portal fibrosis and bile duct proliferation with mild giant cell hepatitis at disease onset with occasional intraductal cholelithiasis[9]. MDR3 immunohistochemical staining will be absent, decreased, or potentially normal if there are functional protein defects[9]. Carcinogenesis and the development of both cholangiocarcinoma and hepatocellular carcinoma have been reported[9,60,61].
Expanded PFIC
TJP2(TJP2):Recently, alternate proteins have been identified in whom mutations result in a phenotypic pattern that is similarly to ‘’classic’’ PFIC disease, mainly cholestasis presenting in the neonatal period. The first of these identified stems from loss of function mutations inTJP2encoding the tight junction protein TJP2. TJP2 is one of the intracellular anchors for tight junctions that seal canaliculi and prevent damage from cytotoxic detergent bile salts[4,59]. To date the largest case series consists of 12 infants from 8 families (most consanguineous) who presented ≤ 3 mo of age with severe liver disease[62]. Though still exceedingly rare, advances in genetic understanding has enabled retrospective re-classification suggesting TJP2 deficiency may be more common than previously thought[63]. The disease results from biallelic mutations inTJP2with extrahepatic manifestation in the respiratory and neurologic systems having been reported. The mechanism of injury is thought to relate to TJP2’s function maintaining junction integrity, the disturbance of which enables toxic molecules to reflux into the paracellular space; however, this is not clearly described[62]. Though few samples are available, pathology demonstrates intracellular cholestasis and giant cell transformation, with absence of TJP2 specific staining[4].Several mutations have been noted specific to the families who manifested the disease, but it is not yet clear if some mutations pertain to less severe disease than others or if there is a milder form of disease that may be appreciated in adult patients.Hepatocellular carcinoma has been described at presentation in infants[64,65]. Due to the severity of presentation, 9 of the initial 12 patients described underwent liver transplant; 2 have survived with portal hypertension, and one passed away of their disease[62].
NR1H4(FXR):PFIC phenotype can also result from mutations inNR1H4, which encodes the FXR, the nuclear receptor transcription factor which regulates BSEP expressionvianegative feedback loop and induces FGF19 to repress bile acid synthesis[4,66]. Patients reported with these defects are extremely rare, with only 5 patients reported in the literature[67,68]. Without appropriate regulation of BSEP,patients with this defect have presented in the neonatal period with normal GGT cholestasis, normal liver enzymes, elevated serum bile acids, extremely elevated AFP,and rapidly progressed to end stage liver disease with vitamin K independent coagulopathy and hyperammonemia[67,68]. On native liver pathology, the patients were found to have intralobular cholestasis with ductular reaction, hepatocellular ballooning, giant cell transformation, and fibrosis with progression to micronodular cirrhosis. Three patients underwent liver transplant with 2 of 3 showing steatosis in the graft organ on follow up[67].
MYO5B(MYO5B):Defects inMYO5B, on which BSEP depends to localize to thecanalicular hepatocellular membrane, usually cause microvillus inclusion disease but also may result in isolated liver disease[4]. Without appropriate BSEP localization,secretion of bile acids is impaired and causes hepatocellular toxicity[69]. This results in a clinical picture of low GGT cholestasis, hepatomegaly, normal or mildly elevated transaminases. Patients have preserved synthetic function but struggle with pruritis and present around 1 year of age, similar to FIC1 and BSEP disease[69]. The hepatocellular damage results in a pathologic pattern of hepatocellular cholestasis with portal and lobular fibrosis and giant cell transformation. Present but abnormal BSEP and MDR3 staining suggest that these transporters are made but can’t appropriately migrate to the canalicular membrane[69].
Because MYO5B interacts with rab11 for appropriate functioning of polarized cells,extrahepatic manifestations can be present. MYO5B has previously been implicated in microvillous inclusion disease, thus some patients with genetic cholestasis have also had diarrheal manifestations of disease[69]. Similarly, some patients also suffer short stature, though others have normal growth. Finally, some patients with this disease have neurologic findings, though it is not clear if these are related to the gene defect[4,69]. In addition to supportive care for nutrition and diarrhea, patients have been treated with antipruritic and anticholestatic agents, including UDCA, rifampin,cholestyramine, traditional Chinese medicine[4,69]. If pruritis is refractory to medical therapy, some success has been seen with PEBD. Finally, liver transplant has been undertaken if pruritis is refractory, though it does not address extrahepatic symptoms[69]. At our institution, the association between MYO5B defects, intestinal failure, and isolated liver disease has made decisions regarding type of transplant(isolated bowel, liver bowel, multi-visceral,etc) challenging in patients with microvillus inclusion disease.
USP53(USP53) and LSR(LSR):A recent report utilizing exome sequencing and positional mapping was able to identify 2 novel loci with defects associated with low-GGT cholestatic liver disease presenting in childhood[70]. In the first case, 3 members of a family (2 sisters and a cousin) presented with low-ggt cholestasis, liver enzyme elevations, and pruritus. Defects in the USP53 protein, thought to colocalize with TJP2 and be part of the tight junction complex[71], was identified. In the second case, a young boy who presented with hypocalcemic seizures, pruritus, liver enzyme elevation, and low-ggt cholestasis was found to have a mutation in lipolysisstimulated lipoprotein receptor (LSR). Mechanisms by which LSR contributed to the liver disease were not reported, although LSRs role in animal models of liver development suggests an area for future research[70].
Contributions beyond pediatrics
The traditional understanding of the PFIC-associated genes contributing to morbidity in adults mainly encompass the phenotypes of BRIC and ICP. The phenotype of BRIC is characterized by intermittent episodes of cholestasis with varying degrees of severity. BothATP8B1andABCB11mutations have been associated with the phenotype[12,51]. While classic descriptions of BRIC note complete symptom resolution without progression, several cases have been reported to transition to more persistent,progressive disease[72]. Treatment of cholestatic episodes with steroids, choleretic agents, and bile acid binders have generally been ineffective, although rifampicin has been shown to decrease pruritus and shorten exacerbations[73,74]. ICP is a common condition affecting about 1% of all pregnancies[75]. ICP manifests during pregnancy with pruritus, hepatic impairment, and cholestasis which usually resolves completely after delivery. While generally considered benign for the mother, adverse perinatal outcomes for the child, such as fetal distress, premature birth, and stillbirth, can occur[75]. While rare, stillbirth has been shown to be associated with bile acid concentrations of ≥ 100 μmol/L highlighting the importance of close monitoring[76].The use of ursodiol has been shown to symptomatically improve pruritus and decrease the risk of premature birth[77,78]. An expanded understanding of the genetics associated with ICP has identified mutations inABCB4, ABCB11, ATP8B1, ABCC2(associated with Dubin-Johnson), andTJP2contributing to disease[79]. Additionally,variations inNR1H4may be implicated in ICP, possiblyviadownregulation of BSEP expression[80]. Beyond BRIC and ICP, drug-induced injury has been historically been linked to PFIC gene associated polymorphisms[52].
More recently, investigators have begun looking more broadly at the contributions that these genes may have on morbidity in adult populations(Table 2). Mutations inATP8B1,ABCB11,ABCB4, andTJP2have been reported in adults with cryptogenic cirrhosis[81]whileABCB4defects have been linked to the development of sclerosing cholangitis, biliary cirrhosis, and low-phospholipid cholelithiasis[82,83]. Genetic sequencing of large cholestatic populations have revealed disease causing mutations in up to a third of patients, with common variants detected in a high number of thosewithout known disease-causing defects suggesting that they still may have a contributing role to the development of cholestasis[83]. Importantly, several of the recently identified contributing genes, such asNR1H4,MYO5B,USP53andLSRwere not tested for in these studies, suggesting the burden may still be higher.
DISCUSSION
PFIC is a heterogeneous cohort of diseases that present both diagnostic and treatment challenges for clinicians. While significant advancement in bile transport physiology has been made by studying these diseases, the breadth of phenotypes from neonates to adults demonstrates that there remains much more to be understood. In the future,precise molecular diagnosis may allow individualized therapy through gene replacement or protein augmentation therapies.

Table 2 Adult manifestations of progressive familial intrahepatic cholestasis gene mutations
ARTICLE HIGHLIGHTS
Research background
Progressive familial intrahepatic cholestasis (PFIC) is an umbrella term originally used to describe 3 classic genetic-based cholestatic diseases in children. Recent advancements in how genetic defects in proteins affect bile acid homeostasis and caused disease has led to an expanded list of syndromes categorized as PFIC and a growing understanding of how adults can be affected. In this report, we review the literature to summarize the understanding of ‘classic’PFIC diseases and present up-to-date information the expanding list of genetic defects that are now known to contribute to the PFIC phenotype.
Research motivation
Bile acid metabolism, homeostasis, and transport is a complex physiologic process, the importance of which is underscored when defects in the system cause disease. While recent advancements have identified critical genes and protein products that, when defective,contribute to disease, phenotypic variability persists, and treatment remains mainly supportive.Furthermore, it is clearly that additional genes and proteins are likely to be identified as the field continues to evolve. In the future, better diagnostics and precise molecular defect identification may identify individualized therapy options that will improve the care provided to these patients.
Research objectives
The objectives of this work were to thoroughly review the current published literature and present an up-to-date summarization of both the ‘’Classic’’ and ‘’Expanded’’ PFIC diseases.
Research methods
A Medline/PubMed search was performed to identify established articles relating to PFIC as well as reports of defects in PFIC-related genes contributing to morbidity in adult and pediatric populations. Data was manually extracted on disease characteristics. Associated phenotypes with other diseases relating to specific genetic defects were also collected. Treatment strategies were summarized. Data was collated and presented in text, figure, and table format.
Research results
We present a comprehensive summary of the ‘’Classic’’ PFIC disorders resulting from defects inATP8B1(FIC1 protein),ABCB11(BSEP protein), andABCB4(MDR3 protein). We further explore and summarize the “Expanded” PFIC disorders including those related toTJP1(TJP2 protein),NR1H4(FXR protein),MYO5B(MYO5B protein,USP53(USP53 protein), andLSR(LSR protein)defects. While many of these disorders have historically affected children, we also looked to present the growing literature related to the significant morbidity that these diseases cause in adults.
Research conclusions
In this review, we present a comprehensive summary of the current understanding andmanagement of PFIC-related disorders. The recent identification of the “Expanded” disorders underscores the importance of continued exploration of the genetic basis of bile acid homeostasis. However, idiopathic disease remains a considerable challenge to patients and healthcare professionals suggesting opportunities for further investigation. Future strategies to improve the treatment provided to patients affected by these devastating diseases are also critically needed.
Research perspectives
Since their first description in 1969, the last 50 years has brought dramatic advancements in both the understanding and management of PFIC-related diseases. Still, challenges remain.Continued idiopathic disease suggest improvement in diagnostic strategies are needed and treatment options remain frustratingly small. Variability in both phenotype and response to therapy opens the possibility that specific gene defects or modifiers can identify sub-populations where more personalized approaches can be more affective. Improved disease models, bothin vitroandin vivo, are needed to better understand mechanisms and identify therapeutic strategies. Finally, the growing morbidity linked to defects in PFIC-related genes identified in adults highlights the urgency, but also the opportunity, for future investigation.
猜你喜欢
杂志排行

World Journal of Hepatology的其它文章
- Roles of hepatic stellate cells in acute liver failure: From the perspective of inflammation and fibrosis
- Hepatitis C virus cure with direct acting antivirals: Clinical,economic, societal and patient value for China
- Hepatitis C virus antigens enzyme immunoassay for one-step diagnosis of hepatitis C virus coinfection in human immunodeficiency virus infected individuals
- Carvedilol vsendoscopic variceal ligation for primary and secondary prevention of variceal bleeding: Systematic review and metaanalysis
- Neonatal cholestasis and hepatosplenomegaly caused by congenital dyserythropoietic anemia type 1: A case report
- Successful treatment of noncirrhotic portal hypertension with eculizumab in paroxysmal nocturnal hemoglobinuria: A case report