木论喀斯特常绿落叶阔叶混交林土壤细菌多样性及其最优采样数
2019-06-04宋同清曾馥平彭晚霞
陈 莉,宋同清,,*,王 华,,曾馥平,,彭晚霞,,杜 虎,,苏 樑
1 中国科学院亚热带农业生态研究所亚热带农业生态过程重点实验室,长沙 410125 2 湖南农业大学生物科学技术学院,长沙 410128 3 中国科学院环江喀斯特生态系统观测研究站,环江 547100
微生物是地球上现存多样性最多的生命形式,其分布也最为广泛,几乎涉及到地球上的所有生境[1-2]。土壤微生物是陆地生态系统中最活跃的成分,是退化生态系统恢复的“先锋者”,它推动着生态系统的能量和物质循环,被公认为土壤生态系统变化的预警及敏感指标[3-4]。而细菌在土壤微生物中占居绝对优势[5-8],有时甚至高达95%以上[3],其所占比例受土层深度、土壤理化性质等诸多因素的影响。以往的学者大多注重于农田和草地生态系统的研究,而对森林生态系统中的土壤细菌报道研究较少[9]。大量研究发现,森林生态系统具有极其丰富的微生物资源,其中变形菌是温带森林土壤细菌中含量最丰富的类群[10],而酸杆菌是温带森林土壤细菌中相对丰度最高的类群[8]。随着分子生物学的发展,土壤微生物多样性的研究手段也在不断地改进,以往对于土壤微生物的研究方法有平板培养法、Biolog微平板法、磷脂脂肪酸谱图分析法(PLFA)、基于PCR技术的方法[11]等,但这些方法手段的微生物通量和分辨率均不高,检测的灵敏度也不够。而高通量测序技术(第二代测序技术)无需构建克隆,耗时少,通量高,错误率低[12],使得简单、快速、较准确地获取土壤微生物信息成为可能,为准确揭示微生物种群结构方面的研究提供了强有力的手段[13-15]。
森林土壤细菌或微生物群落调查的第一步是获得一定数量的森林土壤样品,且这些样品能够代表它们所在的群落。目前的研究中每样地或样方并没有统一的土壤样品的采集数量。那么究竟多少个土壤样品才能充分反映森林土壤微生物群落的信息呢?这是一个基本且重要的问题,同时也是一个需要进一步研究的问题。
广西木论喀斯特常绿落叶阔叶混交林是目前世界上保存最好、面积最大、代表性最强的喀斯特森林集中连片区,生物环境类型特殊,物种组成丰富,群落结构复杂,是研究喀斯特地质背景下生态学问题的理想场所。本文基于Illumina HiSeq测序平台对位于木论国家级自然保护区典型森林(常绿落叶阔叶混交林)内的25 hm2大样地土壤细菌多样性进行了初步研究,分析了木论大样地土壤细菌多样性水平以及土壤细菌的门、纲、目、科、属等不同分类水平的优势类群和各类群相对丰度,以揭示取样数量对土壤细菌多样性检测结果的影响,其结果结果为认识生态功能提供理论知识,将有助于土壤细菌多样性及其与植物多样性的关系等进一步研究,对指导喀斯特退化生态系统植被的迅速恢复和生态重建具有重要意义,同时也可为土壤微生物生物地理学的发展提供科学理论支持和参考。
1 材料与方法
1.1 研究区概况与土壤样品采集
木论国家级自然保护区位于广西环江毛南族自治县西北部,地理坐标为25°07′01″—25°12′22″N,107°54′01″—108°05′51″E,南缘云贵高原,北连贵州茂兰国家级自然保护区,总面积8969 hm2,海拔400—1000 m。该区域为中亚热带季风气候,年平均气温15—18.7℃,最冷月(1月)平均气温3.4—8.7℃,最热月(7月)平均气温23—26.7℃,年积温4700—6300℃;年降雨量1530—1820 mm,集中于4—8月,年平均相对湿度为80%—90%,无霜期235—290 d,气候温暖,雨量充沛。土壤主要为石灰土和零星分布的硅质土,河流及水时空分布不均,地域性来水差异大。研究区植被为中亚热带石灰岩常绿落叶阔叶混交林,属隐域性喀斯特森林植被顶极群落类型,是目前世界上同纬度地区残存下来的仅有的、原生性强、相对稳定的喀斯特森林生态系统,也是喀斯特区原生性森林分布面积最大的区域,植被成层现象比较明显。
1.2 样品采集
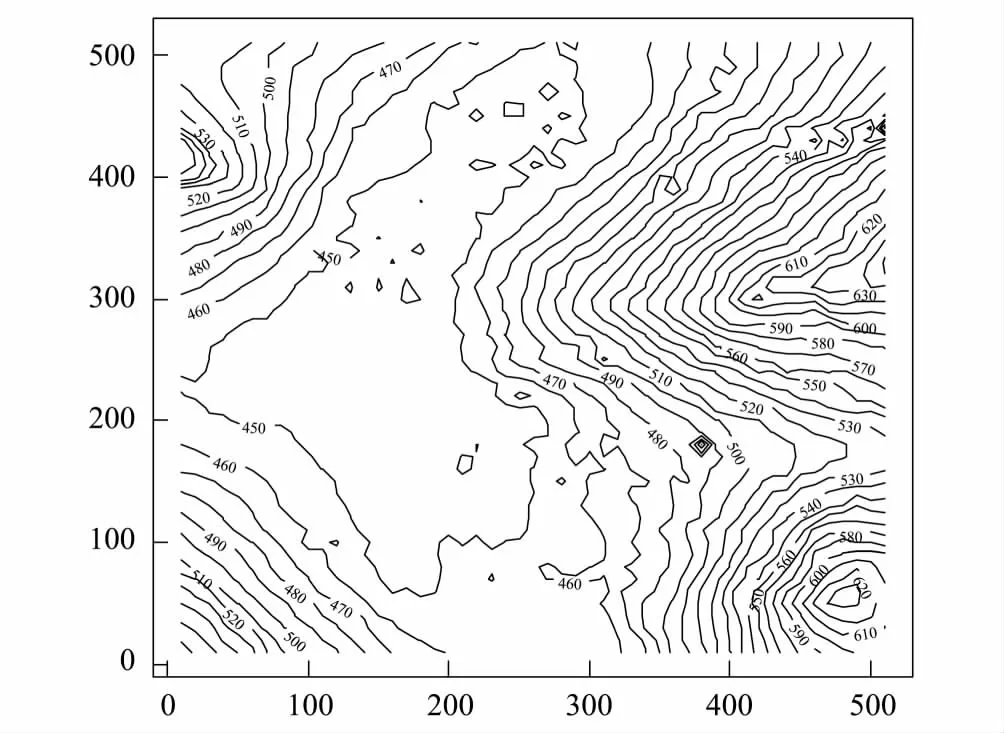
图1 木论样地地形图Fig.1 Thecontour map of the 25 hm2 Mulun forest plot
通过勘测,于2014年3月在研究区内设置了一块500 m×500 m的大样地,作为长期监测样地(图1)。参照CTFS标准采用规则网格法(20 m×20 m)进行样点布置和采样,用全站仪和GPS基站相结合的测量方法设置和划分样地,参考木论自然保护区的森林状况和植被、地形分布状况,共设置625个小样方,样方4个角用水泥桩作永久标记。将纵横各5个20 m×20 m的小样方作为1个100 m×100 m的大样方,在样方的4个角和中心的小样方内进行采样,共计85个样方。采样时在每个样点周围2 m范围内随机采取5个0—10 cm表层土壤混合成一个样品,代表该样点土样,并混合均匀、去除根和石头,-80℃保存用于DNA的提取和后续分析(由于采样过程中样品丢失,因此最后样品数量为82个)。
1.3 DNA提取、扩增和高通量测序
土壤微生物宏基因组DNA提取采用的专用试剂盒(FastDNA®SPIN Kit for Soil,MP),采用引物515F(GTGCCAGCMGCCGCGGTAA)和806R(GGACTACHVGGGTWTCTAAT)对16S rRNA基因的V4高变区进行PCR扩增。PCR反应体系30 μL:15 μL Phusion Master Mix(2×),3 μL(6 μmol/L) Primer(2 μmol/L),10 μL(5—10 ng)gDNA(1 ng/μL),2 μL H2O。PCR反应条件为:98 m℃预变性1 min;30个循环包括(98℃,10 sec;50 m℃,30 sec;72 m℃,30 sec);72℃,5 min。依据PCR产物浓度,将所有扩增成功的PCR产物等量混合,再经定量等质量控制后,将质量合格的PCR产物进行DNA文库的构建,采用Illumina Hiseq平台进行双末端250 bp测序(委托诺禾致源生物信息科技有限公司完成)。
1.4 高通量测序数据处理与统计分析
根据Barcode序列和PCR扩增引物序列从下机数据中拆分出各样品数据,截去Barcode和引物序列后使用FLASH(V1.2.7,http://ccb.jhu.edu/software/FLASH/)[16]对每个样品的reads进行拼接,过滤掉低质量的序列。使用Qiime(V1.7.0,http://qiime.org/scripts/split_libraries_fastq.html)[17]的Tags质量控制流程做聚类及多样性分析。其中,利用Uparse软件(Uparse v7.0.1001,http://drive5.com/uparse/)[18]对所有样品进行聚类,依据惯例[19]以97%的一致性将序列聚类成为OTUs(Operational taxonomic units),依据其算法原则,筛选出OTUs中出现频数最高的序列作为OTUs的代表序列,用RDP[20]对其进行系统分类。再用QIIME[17]进行单样品组成分析,得到样品在不同分类水平上的细菌类群组成及相对丰度的数据,将各分类水平:Phylum(门)、Class(纲)、 Order(目)、Family(科)、Genus(属)上最大丰度排名前10的物种,生成物种相对丰度图。
在上述基础上制作采样点数-土壤细菌类群数曲线。具体的做法是:从82个样品中随机抽取1个样品,记下这个样品中包含的细菌类群数目;然后从剩下的81个样品中再随机抽取1个样品数据,和第一个抽到的样品数据合并,记下这2个样品中包含的细菌类群数目;依此类推,直至采样点的数目达到最大:重复99次取平均值,制作采样点数-土壤细菌类群数曲线。
2 结果与分析
2.1 高通量原始数据处理
测序原始数据得到8872996条reads,拼接并进行预处理、质控后剩余6381418条序列。预设每个样品46431条序列,然后去除嵌合体,去除Singleton(只有1条序列的那些OTU),并标准化为每样品37567条序列,得到每个样品的序列数及OTU数(表1)。
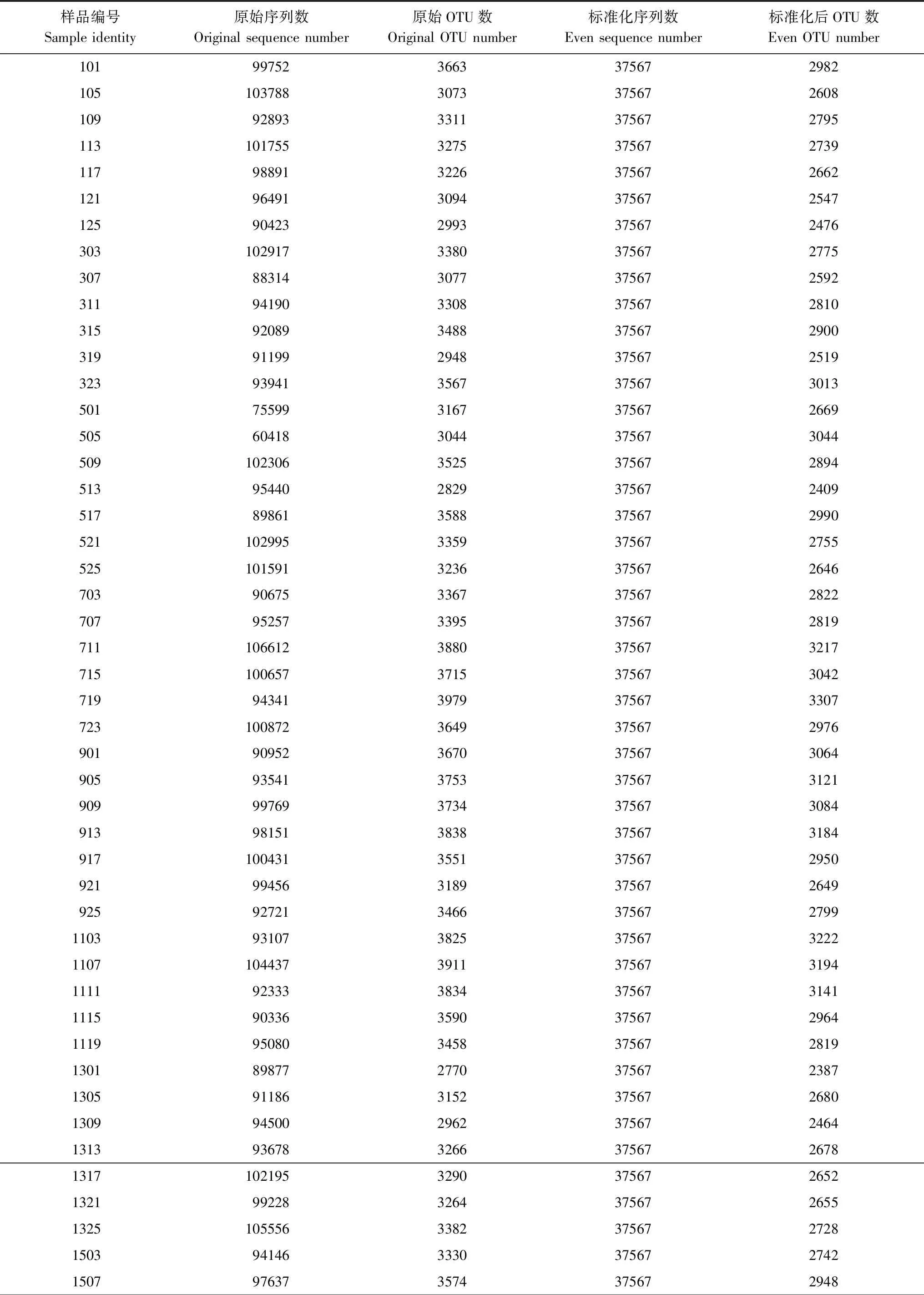
表1 样品序列数及OTU数

续表样品编号Sample identity原始序列数Original sequence number原始OTU数Original OTU number标准化序列数Even sequence number标准化后OTU数Even OTU number151195553315937567258115158903627633756723261519979013038375672548152398611302037567252917011007703402375672760170594554326037567267317099253234493756728551713887483094375672561171792319324637567269017219721033723756727711723924573234375672685190391404300237567245919071041493239375672681191199487302237567246619159197932443756726391919955203066375672507192386903313237567260021018812128533756723622103950353559375673027210993149306437567250821139249233463756727742117103449284837567238721219764529193756724292125100156283337567237323039640132723756726862307875803002375672488231196295301737567249523159238629763756724462319978163104375672545250110096734143756727752505100718318237567259125098261831803756727002513945492947375672490251710242334293756728052521987773190375672640
2.2 土壤细菌分类
对木论样地82个土壤样品进行高通量测序,共产生269822个OTU。未获得注释信息的OTU约占总数的0.064%。82个土壤样品的平均OTU为(3290.51±290.28)个,其中单个样品最多的有3979个、最少的有2763个,分属于44门99纲180目356科630属。
2.2.1门
82个土壤样品平均为细菌28.8±2.6门,其中单个样品最多的有36门,最少的有23门。相对丰度最高的5个门所占比例总和达到87.67%,分别是变形菌门(Proteobacteria)(34.51%)、放线菌门(Actinobacteria)(30.74%)、酸杆菌门(Acidobacteria)(12.24%)、绿弯菌门(Chloroflexi)(6.01%)和奇古菌门(Thaumarchaeot)(4.17%)。
2.2.2纲
82个土壤样品平均有细菌60.9±3.8纲,其中单个样品最多的有70纲,最少的有53纲。相对丰度最高的5个纲所占比例总和达到59.50%,分别是α-变形菌纲(Alphaproteobacteria)(18.43%),嗜热油菌纲(Thermoleophilia)(11.57%)、未确认酸杆菌纲(unidentified-Acidobacteria)(11.10%)、未确认放线菌纲(unidentified-Actinobacteria)(10.65%)和δ-变形菌(Deltaproteobacteria)(7.75%)。
2.2.3目
82个土壤样品平均有细菌108.1±6.0目,其中单个样品最多的有123目,最少的有92目。相对丰度最高的5个目所占比例总和达到37.75%,分别是根瘤菌目(Rhizobiales)(12.50%)、Gaiellales(8.48%)、Subgroup-6(6.63%)、酸微菌目(Acidimicrobiales)(6.48%)和黄色单胞菌目(Xanthomonadales)(3.66%)。
2.2.4科
82个土壤样品中平均有细菌(206.8±11.6)科,其中单个样品最多的有236科,最少的有185科。相对丰度最高的5个科所占比例总和达到14.96%,分别是黄色杆菌科(Xanthobacteraceae)(3.54%)、小单孢菌科(Micromonosporaceae)(3.49%)、0319-6A21(3.04%)、Gaiellaceae(2.76%)和生丝微菌科(Hyphomicrobiaceae)(2.13%)。
2.2.5属
82个土壤样品中平均有细菌(239.6±21.0)属,其中单个样品最多的有322属,最少的有203属。相对丰度最高的5个属所占比例总和为9.21%,说明细菌的属水平的多样性不显著。相对丰度最高的5个属分别是Gaiella(2.76%)、小单孢菌属(Micromonospora)(1.65%)、土微菌属(Pedomicrobium)(1.62%)、溶杆菌属(Lysobacter)(1.60%)和链霉菌属(Streptomyces)(1.55%)。
2.3 取样数量对土壤细菌多样性的影响
在不同的分类水平上,随着取样点的增加,检测出的土壤细菌类群数目也在增加。当取样点达到82个时,土壤细菌类群的数目达到最大,分别为44门99纲180目356科630属269822个OTU(图2)。
在门的分类水平上,取样点数在1—6之间时,检测到的细菌门数量随着取样点数量增加而快速增加;取样点数超过6个后,门的数量增速减慢;当取样点数为36个时,约能检测到43个门;取样点数在37—82之间时,门数仅增加1个,此时门数量为达到44。
在纲的分类水平上,取样点数在1—13之间时,检测到的细菌纲数量随着取样点数量增加而快速增加;取样点数超过13后,纲的数量增速减慢;当取样点数为57个时,约能检测到98个纲;取样点数在57—82间时,纲数仅增加1个达到99个。
在目的分类水平上,取样点数在1—31区间时,检测到的细菌目数量随着取样点数量增加而快速增加;取样点数超过31后,目的数量增速减慢;当取样点数为72个时,约能检测到179个纲;取样点数在73—82区间时,目数仅增加1个达到180。
在科的分类水平上,取样点数在1—62区间时,检测到的细菌科数量随着取样点数量增加而快速增加;取样点数超过62后,科的数量增速减慢,但增速仍较大;当取样点数为78个时,约能检测到355个目;取样点数在79—82区间时,科数仅增加1个达到356。
在属的分类水平上,取样点数在1—71区间时,检测到的细菌属数量随着取样点数量增加而快速增加;取样点数超过71后,属的数量增速减慢,但增速仍较大,且属的增速大于科的增速。在OTU的分类水平上,取样点数在1—82区间时,检测到的细菌OTU数量随着取样点数量增加一直在较快增加(图2)。

图2 木论大样地82个土壤样品中检测到的各分类水平上细菌类群数量随采样点数增加的稀疏曲线Fig.2 Rarefaction curves of the observed bacterial quantity at different taxonomic levels varying with number of samples in the Mulun Plot
3 讨论
本文采用Illumina公司Hiseq测序仪对地处中亚热带的木论常绿落叶阔叶混交林土壤细菌多样性进行了初步研究,列出了木论森林土壤壤细菌门、纲、目、科、属、OTU数量以及不同分类水平上的优势类群及其相对丰度。虽然本次调查采集的土壤样品数量有限,但高通量测序技术的应用为进一步调查木论大样地内土壤细菌等微生物资源积累了经验,并提供了新的技术支撑。在本研究中,与数据库比对不上的OTU约占总数的0.064%,这说明所参考的GreenGenes数据库中包含了木论森林土壤细菌类群中的绝大部分。研究显示,木论森林土壤细菌在各分类水平上的信息较明确,这可能与测序区间、序列长度以及比对的数据库等有关,测序区间、序列长度的增加,也有可能检测出来更多的细菌类群。而随着分类水平的降低,所检测出的细菌类群越多,是因为分类水平越低,细菌的类群种类就越丰富,因此土壤细菌相对丰度前10的类群在所有类群中所占的比例也越低。
本研究在木论喀斯特常绿落叶阔叶混交林土壤中共检测出变形菌门、放线菌门、酸杆菌门等44门,变形菌门是最丰富的细菌类群,这一结果也与国内外关于森林、草地以及农田土壤细菌多样性方面的很多研究结果相一致[22-24]。Yuan等[25]对我国青藏草地研究发现酸杆菌是土壤中含量最丰富的细菌类群,Fierer等[8]也发现在温带森林中酸杆菌是土壤中含量最丰富的细菌类群,在对木论大样地森林土壤的研究中,酸杆菌门是仅次于变形菌门和放线菌门居细菌类群总含量第三位,这也证实了杨官品等[26]的观点,在土壤细菌中存在非常丰富的多样性,不同类型的土壤中具备各自的优势细菌类群,而同时又含有相似的细菌类群。
Nacke等[24]通过在德国的研究表明,土壤细菌群落的结构在很大程度上是受树种和土壤pH值影响的;也有学者认为在不同的空间尺度上,pH值对土壤细菌的多样性和群落组成均会产生很大的影响[27-29]。植物多样性、土壤理化性状、土壤温度是形成森林土壤微生物群落结构的3个主要因素[30]。到底是哪些环境因子影响着木论森林土壤细菌群落的组成和结构?土壤细菌不同类群又存在什么样的功能?土壤细菌与其他生物类群之间是如何相互作用的?以及土壤细菌在森林群落维持上扮演着什么样的角色?这些问题都有待进一步的深入探讨和研究。
在已有土壤细菌或土壤微生物研究中采用的取样数量变化较大(如1个、10个、36个样品等),但是具体到某种植被类型、生境类型或是某个生态系统,需要采集分析多少土壤样品才能较好地反映其微生物多样性水平,仍然是一个值得探究的问题。值得注意的是,在采样数量一定的情况下,多数研究还是将数个取样点土样混合后形成混合样品以增加样品的代表性[1,9,31-32]。对木论大样地森林土壤细菌多样性的初步研究表明,取样数量对检测出的土壤细菌类群数量有显著影响:随着样品数量增多,被检测出来的细菌类群数量也增多,当样品达到82个时,其增速虽然变缓但仍保持上升的趋势,且各分类水平上的细菌类群多样性表现出类似趋势的同时也存在一定区别,这表明增加土壤采样点数量应能检测出更多的细菌类群,同时也说明在进行不同研究时要考虑取样数量的影响。本研究结果表明,各分类水平在取样点数一定时,检测到的细菌数量随着取样点数的增加而增加,但增速不同,这说明当取样点数较少时,可能会有较多的细菌类群没有被监测出来。在木论常绿落叶阔叶混交林中,在门的分类水平上,36个样品基本上可代表82个样品监测出来的细菌门多样性信息;在纲的分类水平上,57个样品基本上可代表82个样品监测出来的细菌纲多样性信息;在目的分类水平上,72个样品基本可以代表82个样品监测出来的细菌目多样性信息;在科的分类水平上,78个样品基本可以代表82个样品监测出来的细菌科多样性信息;在属的分类水平上,当样品数量达到82个时检测出来的相应细菌类群数仍在不断的增加;特别是在OTU的水平上,当样品数量达到82个时细菌OTU数量仍呈现出较快增加的趋势,由此推测,如果增加较多样品数量,检测出来的细菌OTU数量还会有较大增加。
获取一定数量的土壤样品是森林土壤微生物群落调查的首要步骤,且这些样品能够代表它们所在的群落。以往的研究中土壤样品的采集有每个样地或样方1个[29-30]、10个[33-34]、36个[9]等不同数量,究竟多少数量土壤样品才能充分反映森林土壤微生物群落的信息,这是一个基本且重要的问题[35],也是一个需要进一步深入探讨与研究的问题。在以往对草地[24-25]、农田[36-37]或是森林[18,38]等的土壤微生物研究中,均考虑取样数量对于研究结果的影响。取样数量除了受研究目的和研究深度决定外,同时也会受到研究成本的影响,特别是在高通量测序单个样品所需经费较高的情况下,因此,在实际研究中需要对于取样数量进行权衡,既要能满足研究目的、反应所研究对象的微生物多样性水平,又要能兼顾到实验的研究费用。另外,文库的构建及测序深度的选择等也会对检测出的细菌类群产生一定程度影响。库容和测序深度的增加,也有可能检测出来更多的细菌类群。还有,测序区间的选择也会对研究结果产生一定的影响,例如:16S rRNA基因的高变区不同,其分类信息的精确性也会不一样[39]。
致谢:感谢木论国家级自然保护区工作人员对野外工作的帮助。
