辣椒溶杆菌Lysobacter capsici X2-3遗传操作体系的建立
2019-06-03王虹李星志赵丹刘爱新
王虹 李星志 赵丹 刘爱新


摘要:辣椒溶杆菌(Lysobacter capsici)X2-3是本实验室从小麦根际土壤中分离的对多种植物病原真菌和卵菌有明显拮抗活性的菌株,建立有效的遗传操作体系对了解其基因的功能具有重要意义。本研究以VgrG为目标基因,比较了不同载体对X2-3基因敲除效果,结果发现,载体pEX18GM、pBR325和pK19mob转化X2-3后,均未得到VgrG缺失突变体;而载体pKMS1转化X2-3后,经10%蔗糖二次筛选、PCR和Southern blot验证等,成功得到VgrG缺失突变体;用广宿主载体pBBR1MCS-5构建的VgrG互补突变体,可以恢复VgrG基因的功能。本结果为进一步研究该菌的基因功能提供了技术保障。
关键词:辣椒溶杆菌;基因敲除;载体筛选;VgrG基因
中图分类号:S476:Q933文献标识号:A文章编号:1001-4942(2019)04-0007-06
Abstract Lysobacter capsici X2-3 was isolated by our lab from wheat rhizosphere soil with antimycotic activities to many plant pathogenic fungi and oomycetes. Establishing an effective genetic manipulation system was of great significance for understanding the function of its genes. Using VgrG gene as target, the study compared gene knockout effect of four different vectors on X2-3. The results showed that X2-3 transformed by the vectors pEX18GM, pBR325 and pK19mob were not VgrG deletion mutates. However, after transformation of vector pKMS1 into X2-3, VgrG deletion mutants were successfully obtained by 10% sucrose secondary screening, PCR and Southern blot verification. The complemented mutants of VgrG constructed by broad-host vector pBBR1MCS-5 also restored the function of VgrG gene. This study provided a technical guarantee for the further study on the gene function of the strain X2-3.
Keywords Lysobacter capsici; Gene knockout; Carrier screening; VgrG gene
植物病害的生物防治方法近年来日益受到人们的重视,生产上已广泛利用的生防菌包括木霉和菌根真菌等真菌[1],链霉菌等放线菌[2],芽孢杆菌、假单胞杆菌等细菌[3]。溶杆菌属(Lysobacter)常存在于水和土壤中,与芽孢杆菌、假单胞杆菌等细菌相比,生长较慢,分离纯化较为困难,因此分离出现的比例相对较低[4-6],研究也相对较少。
辣椒溶杆菌X2-3(以下简称X2-3)是本实验室从小麦根际分离到的一株对真菌、卵菌和革兰氏阳性细菌有显著拮抗活性[7,8]的菌株。研究发现,辣椒溶杆菌菌株可以产生多种胞外酶和抗菌物质[7-9],有着很好的应用前景。但由于缺乏有效的遗传操作体系,对X2-3的抗菌物质及生物合成机制等了解很少。本研究以VgrG基因为目标基因构建不同的敲除载体并转化X2-3,利用平板筛选、PCR、酶切及Southern blot检测等技术手段对敲除VgrG基因的突变体进行验证,同时构建VgrG互补菌株,以期建立X2-3有效的遗传操作体系,并为研究该菌的基因功能提供参考。
1 材料与方法
1.1 试验材料
供试菌株为Lysobacter capsici X2-3;大肠杆菌为Escherichia coli DH5α、E. coli S17-1,均由本实验室保存,所用质粒见表1。供试培养基NA和LA分别用于L. capsici X2-3、大肠杆菌培养。引物(表2)由生工生物工程(上海)股份有限公司合成。
1.2 试验方法
1.2.1 菌株X2-3对不同抗生素的敏感性试验 将菌株X2-3在NB培养液中28℃、180 r/min振荡培养12~24 h,6 000 r/min离心10 min,弃上清,菌体用无菌水调节OD600为1.0。取100 μL菌液涂布于含有不同浓度抗生素的NA培养基,28℃培养12~24 h,观察菌株生长状况。所用抗生素包括庆大霉素(Gm)、卡那霉素(Kna)、氨苄西林(Amp)和四环素(Tet),各抗生素濃度梯度为10、20、50、100、200、500 μg/mL。以不含抗生素的NA为对照。
1.2.2 基因敲除重组载体的构建 用引物VgrG1F/VgrG1R和VgrG2F/VgrG2R,以菌株X2-3基因组DNA为模板分别扩增VgrG基因的上、下游同源臂。PCR反应体系:10×buffer 2.5 μL,2.5 μmol/L dNTP Mixture 2 μL,10 μmol/L 5′端引物1 μL,10 μmol/L 3′端引物1 μL,5 U/μL DNA polymerase 0.5 μL,100 ng/μL DNA模板1 μL,ddH2O 17 μL。PCR扩增程序:94℃ 5 min;94℃ 40 s,65~67℃ 40 s,72℃ 40 s,30个循环;72℃ 10 min,电泳回收同源臂连接至克隆载体pMD19-T Simple Cloning Vector并测序。测序无误后,将VgrG基因的上、下游同源臂与pEX18GM[10]、pPKMS1[11]、pBR325[12]、pK19mob[13]载体用相同的限制性内切酶酶切后,16℃过夜连接,42℃热激转化E.coli S17-1感受态细胞。37℃、180 r/min培养45 min,分别涂布于含有Gm(200 μg/mL)、Kna(100 μg/mL)、Amp(100 μg/mL)、Kna(100 μg/mL)的LA平板,37℃过夜培养。挑取单菌落,进行菌落PCR及酶切验证,鉴于4个载体均具有BamHⅠ、HindⅢ的酶切切点,确定20 μL酶切体系为:10 × K buffer 2 μL,质粒8 μL, 20 U/μL BamHⅠ1 μL,20 U/μL HindⅢ 1 μL,ddH2O 8 μL。最后将验证正确的重组载体转化X2-3感受态细胞。
1.2.3 突变体的筛选及PCR验证 根据载体pEX18GM[10]、pPKMS1[11]、pBR325[12]和pK19mob[13]的抗生素抗性类型,分别用抗生素Gm(200 μg/mL)、Km(500 μg/mL)、Tet(10 μg/mL)和Km(500 μg/mL)对经各重组载体转化的X2-3进行初步筛选。对获得的单菌落根据载体特点进一步分析筛选:载体pEX18GM和pKMS1因含有蔗糖敏感基因SacB,二次筛选用含10%蔗糖的NA培养基,未发生二次交换的交换子将在蔗糖培养基中致死[14];pK19mob中含有gusA基因,用氨基葡萄糖苷為底物的培养基进行二次筛选[13];而载体pBR325在菌株生长过程中可以自然脱落[12,15],无需二次筛选。最后,对获得的单菌落进行PCR验证。
1.2.4 杂交验证 经PCR初步验证,获得缺失突变株。为明确获得的菌株是否为基因缺失突变体,随机挑选PCR结果正确的单菌落提取基因组DNA,进一步用Southern blot进行验证。分别设计探针,并以基因组DNA为模板克隆探针,电泳切胶回收并测序。选择合适的酶切位点对基因组DNA进行定量酶切,参照文献[16]的方法进行杂交验证。
1.2.5 互补菌株的筛选及验证 根据X2-3基因序列设计引物HBVgrGF/HBVgrGR,用高保真酶扩增目的基因VgrG完整的开放阅读框,电泳回收基因VgrG片段后连接至pEASY-Blunt Simple Cloning Vector并测序。测序无误后,与同样酶切的pBBR1MCS-5连接、转化大肠杆菌DH5α,步骤参照1.2.2。对单菌落进行PCR及酶切验证,体系参照1.2.2。将酶切验证正确的重组载体电击转化缺失突变体感受态细胞,并涂布于NA(Gm 200 μg/mL)平板,28℃培养3~5 d。单菌落用PCR等进一步进行验证。
1.2.6 菌株形态观察 取2 μL培养至OD600为1.0的菌液接种于NA平板表面,超净台静置直至菌液固定在培养基上不再流动,28℃静置培养3~5 d。用Leica体视镜对野生菌株、突变体和互补菌株的菌落形态进行观察,并拍照记录。
2 结果与分析
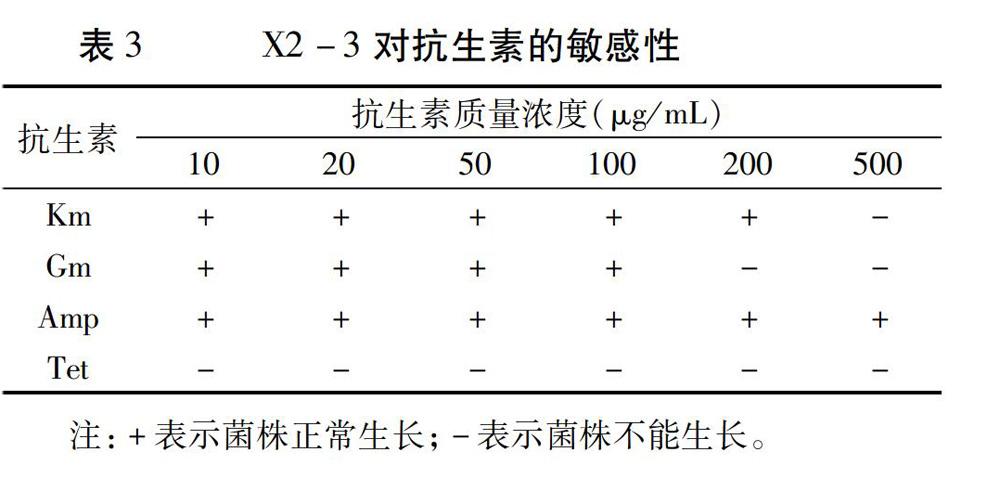
2.1 菌株X2-3 对抗生素的敏感性检测
X2-3在不同抗生素浓度下的生长结果如表3所示。其对氨苄霉素(Amp)敏感性最差,在500 μg/mL浓度下仍可正常生长;对四环素(Tet)最敏感,在10 μg/mL浓度下就不能生长;对卡那霉素和庆大霉素的敏感性居中,在500 μg/mL卡那霉素(Km)和200 μg/mL庆大霉素(Gm)不能生长。本结果为后续突变体筛选时选用合适的抗生素浓度提供了依据。
2.2 不同转化体系比较
以VgrG为目标基因,选用载体pEX18GM、pKMS1、pBR325和pK19mob分别构建VgrG基因的不同敲除载体并转化X2-3,结果(表4)发现,各载体在菌株X2-3中的转化效果存在明显差异。构建的pEX18GM、pKMS1、pBR325和pK19mob重组载体转化后,均可在相应抗生素平板上生长,而野生株与转化空载pEX18GM、pKMS1、pBR325和pK19mob均不生长,表明构建的重组载体均成功转化X2-3菌株。但经含10%的蔗糖培养基和含25 mg/L氨基葡萄糖苷培养基二次筛选时, pEX18GM和pK19mob重组载体转化的菌株均无法正常生长,表明在蔗糖和氨基葡萄糖苷条件下,一次交换子未发生二次交换,故无法生长;转化pBR325重组载体的菌株可以在抗生素的平板上生长,但PCR无法扩增到目的条带,表明pBR325不能用于菌株X2-3中VgrG基因的定向敲除;经重组载体pKMS1-VgrG转化的菌株,可以在10%的蔗糖板生长,经PCR扩增初步分析,可以得到缺失VgrG基因的突变菌株。初步表明载体pKMS1可以用于菌株X2-3的基因敲除。
2.3 基因VgrG敲除重组载体的PCR和酶切验证
构建重组载体pKMS1-VgrG进行PCR验证,将验证正确的用BamHⅠ、HindⅢ酶切,酶切结果见图1。由图1a可见,单菌落1、4、5、6、7均可以清晰地扩增到1 400 bp的条带。以单菌落1的质粒酶切发现,BamHⅠ单酶切可以得到7 200 bp的条带;HindⅢ单酶切可以得到共计7 200 bp的条带,因为载体pKMS1中有2个HindⅢ的酶切位点;双酶切可以得到1 400 bp和载体酶切的条带(图1b),与预期结果一致。这表明基因VgrG敲除重组载体构建成功。
2.4 基因VgrG敲除菌株的筛选
将重组载体pKMS1-VgrG电击转化菌株X2-3感受态细胞,通过抗生素(Km,500 μg/mL)初步筛选、10%蔗糖平板二次交换,用引物VgrG1F/VgrG2R进行PCR扩增,理论上野生株X2-3能扩增到一条2 139 bp的条带,而突变株可以得到1 416 bp的条带。选用蔗糖筛选后的4个单菌落PCR,结果(图2)显示,菌株X2-3扩增到2 139 bp的条带,单菌落3、5可以扩增到1 416 bp的条带,初步确定为敲除成功的突变体,而2、4单菌落未扩增到1 416 bp的条带。
3 讨论与结论
溶杆菌属的细菌分布广泛,可通过分泌胞外酶、小分子化合物及生物表面活性剂和抗生素等物质发挥对多种病原细菌、真菌、线虫等的拮抗性、竞争性、抑制或杀死等作用[17,18],对防治土传病害等具有极大的经济效益和广阔的应用前景[19]。溶杆菌属细菌是一类具有良好潜力的生防细菌,目前,尚无溶杆菌属细菌对人体或者环境有害的报道[20]。与其它种类的细菌相比,其研究相对较少,因此建立一种适用于不同溶杆菌的分子操作手段尤为重要。本研究比较了4种常用自杀载体pEX18GM、pBR325、pK19mob和pKMS1对X2-3的基因敲除效果,发现仅有pKMS1可用于X2-3的目标基因敲除。载体pBBR1MCS-5可在X2-3中用于基因互补。
常用的基因編辑技术包括转座子插入[21]、CRISPR-Cas9[22]、基因敲除[14,23]等,而相较于点突变、插入突变而言,基因敲除可以避免基因重叠对其上下游基因表达的影响,并可以敲除完整的目标基因进而研究其功能,现已发展成为最佳的微生物菌剂改良途径。目前,细菌中常利用同源重组的原理借助自杀载体来完成对基因的定向敲除。产酶溶杆菌(L. enzymogenes)OH11是研究较为深入的一种溶杆菌,前人利用载体pEX18GM对L. enzymogenes菌株OH11中多个基因进行敲除,为了解OH11中基因的功能提供了很多便利[4,9,10]。然而,溶杆菌对不同载体常表现出选择性,目前为止还未有一种适用于所有溶杆菌的基因敲除操作体系。本试验证实产酶溶杆菌OH11中频繁使用的pEX18GM在辣椒溶杆菌X2-3中无法发挥基因敲除的作用。前人研究的载体pJQ200SK在菌株L. gummosus OH17中可有效完成基因敲除[24],但在OH11中则没有效果[25]。可见建立适合各菌株的遗传操作体系是深入研究基因功能的前提。本研究中,我们成功建立起可用于X2-3菌株的遗传操作体系,为下一步研究其基因功能等方面提供了技术保障。
参 考 文 献:
[1] Das M M, Haridas M, Sabu A. Biological control of black pepper and ginger pathogens, Fusarium oxysporum, Rhizoctonia solani and Phytophthora capsici, using Trichoderma spp[J]. Biocatalysis and Agricultural Biotechnology,2019,1(17):177-183.
[2] Deng Q, Xiao L, Liu Y,et al. Streptomyces avermitilis industrial strain as cell factory for Ivermectin B1a production[J]. Synthetic and Systems Biotechnology,2019,4(1):34-39.
[3] Fu Y, Yu Z, Liu S, et al. c-di-GMP regulates various phenotypes and insecticidal activity of Gram-positive Bacillus thuringiensis[J]. Frontiers in Microbiology, 2018, 9:45.
[4] 刘轶儒, 徐菲菲, 钱国良,等. 产酶溶杆菌rpfG基因的克隆与功能分析[J].南京农业大学学报,2013,36(2):45-50.
[5] El-Tarabily K A, Nassar A H, Hardy G E, et al. Plant growth promotion and biological control of Pythium aphanidermatum, a pathogen of cucumber, by endophytic actinomycetes[J].Journal of Applied Microbiology,2009,106(1):13-26.
[6] Zúiga A, Poupin M J, Donoso R et al. Quorum sensing and indole-3-acetic acid degradation play a role in colonization and plant growth promotion of Arabidopsis thaliana by Burkholderia phytofirmans PsJN[J]. Mol. Plant-Microbe Interact., 2013, 26(5):546-553.
[7] 刘朝霞.辣椒溶杆菌X2-3抗菌物质的初步分离及其特性分析[D].泰安:山东农业大学,2016.
[8] Yi J L, Wang J, Li Q, et al. Draft genome sequence of the bacterium Lysobacter capsici X2-3, with a broad spectrum of antimicrobial activity against multiple plant-pathogenic microbes[J]. Genome announcements, 2015, 3 (3):e00589-15.
[9] Qian G, Wang Y, Qian D,et al. Selection of available suicide vectors for gene mutagenesis using chiA (a chitinase encoding gene) as a new reporter and primary functional analysis of chiA in Lysobacter enzymogenes strain OH11[J]. World J. Microbiol. Biotechnol., 2012,28(2):549-557.
[10]Qian G, Xu F, Venturi V, et al. Roles of a solo LuxR in the biological control agent Lysobacter enzymogenes strain OH11[J]. Phytopathology,2014,104(3):224-231.
[11]Zou L F , Li Yu R, Chen G Y . A non-marker mutagenesis strategy to generate poly-hrp gene mutants in the rice pathogen Xanthomonas oryzae pv. oryzicola[J]. Agricultural Sciences in China, 2011, 10(8):1139-1150.
[12]Prentki P, Karch F, Iida S, et al. The plasmid cloning vector pBR325 contains a 482 base-pair-long inverted duplication[J]. Gene, 1981, 14(4):289-299.
[13]Katzen F, Becker A, Verónica Ielmini M, et al. New mobilizable vectors suitable for gene replacement in gram-negative bacteria and their use in mapping of the 3' end of the Xanthomonas campestris pv. campestris gum operon[J]. Appl. Environ. Microbiol., 1999, 65(1):278-282.
[14]Tan Y,Xu D, Li Y , et al. Construction of a novel sacB -based system for marker-free gene deletion in Corynebacterium glutamicum[J]. Plasmid, 2012, 67(1):44-52.
[15]Chen K C , Ravichan dran A , Guerrero A , et al. The Burkholderia contaminans MS14 ocfC gene encodes a xylosyltransferase for production of the antifungal occidiofungin[J]. Applied and Environmental Microbiology, 2013, 79(9):2899-2905.
[16]錢栋宇. 产酶溶杆菌OH11菌株几丁质酶基因的功能研究[D].南京:南京农业大学,2011.
[17]Puopolo G , Cimmino A , Palmieri M C , et al. Lysobacter capsici AZ78 produces cyclo(L-Pro- L-Tyr), a 2,5-diketopiperazine with toxic activity against sporangia of Phytophthora infestans and Plasmopara viticola[J]. Journal of Applied Microbiology, 2014, 117(4):1168-1180.
[18]Xie Y, Wright S, Shen Y, et al. Bioactive natural products from Lysobacter[J]. Nat. Prod. Rep.,2012, 29(11):1277-1287.
[19]Hayward A C,Fegan N, Fegan M,et al.Stenotrophomonas and Lysobacter: ubiquitous plant-associated gamma-proteobacteria of developing significance in applied microbiology[J]. Journal of Applied Microbiology, 2009,108(3):756-770.
[20]Qian G, Hu B, Jiang Y, et al. Identification and characterization of Lysobacter enzymogenes as a biological control agent against some fungal pathogens[J]. Agricultural Sciences in China, 2009, 8(1):68-75.
[21]牟蕾,衣静莉,李星志,等.一株高拮抗活性菌株的鉴定及其抗菌活性缺失突变体的获得[J].山东农业科学,2018,50(7):126-132.
[22]信欣,陈丽杰,薛闯.CRISPR-Cas9技术在细菌中的研究进展及应用[J].微生物学杂志,2018,38(6):97-102.
[23]Quandt J, Hynes M F. Versatile suicide vectors which allow direct selection for gene replacement in Gram-negative bacteria[J]. Gene, 1993, 127(1):15-21.
[24]刘琳琳,钱国良,刘凤权.胶状溶杆菌OH17菌株遗传操作系统的构建[J].南京农业大学学报,2017,40(4):649-654.
[25]钱国良. 产酶溶杆菌生防相关基因的克隆、功能分析及工程菌构建[D].南京:南京农业大学,2009.
