Large-scale Identification and Time-course Quantification of Ubiquitylation Events During Maize Seedling De-etiolation
2019-06-03YueFengWangQingChaoZheLiTianCongLuHaiYanZhengCaiFengZhaoZhuoShenXiaoHuiLiBaiChenWang
Yue-Feng Wang, Qing Chao, Zhe Li, Tian-Cong Lu,Hai-Yan Zheng, Cai-Feng Zhao, Zhuo Shen, Xiao-Hui Li*,Bai-Chen Wang*
1 Photosynthesis Research Center, CAS Key Laboratory of Photobiology, Institute of Botany, Chinese Academy of Sciences,Beijing 100093, China
2 Crop Germplasm Resources Institute, Jilin Academy of Agricultural Sciences, Gongzhuling 136100, China
3 Precision Scientific (Beijing) Co., Beijing 100085, China
4 Advanced Biotechnology and Application Research Center, School of Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing 100083, China
5 Center for Advanced Biotechnology and Medicine, Biological Mass Spectrometry Facility, Rutgers University, Piscataway,NJ 08855, USA
6 College of Life Sciences, Graduate University of Chinese Academy of Sciences, Beijing 100049, China
Abstract The ubiquitin system is crucial for the development and fitness of higher plants.De-etiolation, during which green plants initiate photomorphogenesis and establish autotrophy, is a dramatic and complicated process that is tightly regulated by a massive number of ubiquitylation/de-ubiquitylation events. Here we present site-specific quantitative proteomic data for the ubiquitylomes of de-etiolating seedling leaves of Zea mays L. (exposed to light for 1, 6, or 12 h)achieved through immunoprecipitation-based high-resolution mass spectrometry (MS). Through the integrated analysis of multiple ubiquitylomes,we identified and quantified 1926 unique ubiquitylation sites corresponding to 1053 proteins.We analyzed these sites and found five potential ubiquitylation motifs,KA,AXK,KXG,AK,and TK.Time-course studies revealed that the ubiquitylation levels of 214 sites corresponding to 173 proteins were highly correlated across two replicate MS experiments, and significant alterations in the ubiquitylation levels of 78 sites (fold change >1.5) were detected after de-etiolation for 12 h. The majority of the ubiquitylated sites we identified corresponded to substrates involved in protein and DNA metabolism, such as ribosomes and histones.Meanwhile,multiple ubiquitylation sites were detected in proteins whose functions reflect the major physiological changes that occur during plant de-etiolation,such as hormone synthesis/signaling proteins,key C4 photosynthetic enzymes,and light signaling proteins.This study on the ubiquitylome of the maize seedling leaf is the first attempt ever to study the ubiquitylome of a C4 plant and provides the proteomic basis for elucidating the role of ubiquitylation during plant de-etiolation.
KEYWORDS Maize seedling leaf;De-etiolation;Ubiquitylation;C4 photosynthetic enzymes;Ribosome
Introduction
De-etiolation is a rapid process, through which plants transform into an autotrophic state. Light signal transduction during de-etiolation depends on the ubiquitin-targeted degradation of specific proteins antagonizing photomorphogenesis. Green plants perceive different spectra of light through the photoreceptors phytochrome, cryptochrome, and UV resistance locus 8 (UVR8) [1,2]. Upon the perception of light, seedling de-etiolation initiates with the degradation of photomorphogenesis repressors via ubiquitin modification and the 26S proteasome [3]. Constitutive photomorphogenic 1(COP1),a RING E3 ubiquitin ligase,is the core negative regulator of photomorphogenesis[4].In the dark,the COP1-suppressor of phytochrome A (SPA) complex exerts E3 ligase activity and targets positive regulators of photomorphogenesis,including ELONGATED HYPOCOTYL 5(HY5),LONG AFTER FAR-RED LIGHT1(LAF1),LONG HYPOCOTYL IN FAR-RED 1 (HFR1), B-BOX4/CONSTANS-LIKE3(BBX4/COL3), B-BOX22, and a GATA-type transcription factor GATA2, for degradation via the 26S proteasome [5,6].Phytochrome-interacting factor (PIF) 1, PIF3, PIF4, and PIF5 work as negative regulators in concert with COP/SPA proteins in the dark, whereas PIFs are rapidly ubiquitylated and degraded in the light [5].
Aside from modulating these ubiquitylation-degradation events in light signaling pathways, phytochrome also targets multiple transcription factors at an early stage of light perception,which can subsequently lead to large-scale remodeling of the transcriptome[7].Meanwhile,light signals are transmitted to a complex network of multiple hormone synthesis and signaling pathways to direct de-etiolation [8]. Many hormonal signal transduction pathways share the same regulatory mechanism as the light signaling pathway,i.e.,ubiquitin targets negative factors for degradation.For example,the degradation of the auxin/indole-3-acetic acid (AUX/IAA) proteins in the auxin signaling pathway enhances auxin signaling and promotes phototropism[9].Asp-Glu-Leu-Leu-Ala(DELLA)proteins are targeted by ubiquitination and degradation to activate the gibberellin signaling pathway, which represses photomorphogenesis in the dark, and similarly,JASMONATE-ZIM DOMAIN (JAZ) proteins are targeted for ubiquitin-mediated degradation to activate the jasmonate signaling pathway, which regulates plant development in response to light [10,11].
In response to light and hormonal signals, greening of the etiolated leafis evident within 12 h [12]. Pigments needed for photosynthesis, including chlorophylls and carotenoids, are rapidly synthesized in large amounts[13].Meanwhile,chromosome and nucleosome architecture is reorganized [14]. These physiological changes during de-etiolation are all orchestrated by ubiquitin-mediated signaling events.Thus,it is important to study ubiquitylation at the proteomics level to comprehensively understand the de-etiolation process.
Over the years, there have been many reports on the ubiquitylomes of plants,including Arabidopsis thaliana,rice(Oryza sativa), and common wheat (Triticum aestivum L.) [15-18].Our understanding of the ubiquitylation targets in C3 plants has greatly increased in the past six years. Studies have indicated that the ubiquitin-proteasome pathway is the dominant system for selective protein turnover in plants [19,20]. Variation in ubiquitin (Ub) modification also facilitates the regulation of protein subcellular localization, ribosomal protein synthesis,chromatin structure, DNA damage repair,cell cycle progression, apoptosis, and transportation [20-23]. However,there are few reports on ubiquitylated proteomes in crops.
To fill this gap in knowledge and facilitate detailed investigations on the role of ubiquitylation during de-etiolation,here we established a dynamic ubiquitylation profile for deetiolating seedling leaves of maize inbred line B73.Immunoprecipitation-based proteomic analysis coupled with large-scale mass spectrometry (MS) [24] was conducted to identify and quantify ubiquitylation sites. Using these ubiquitylome data, we provide a time-course atlas of ubiquitylation targets in the maize leaf during the first 12 h of the deetiolation process.
Results and discussion
Identification of ubiquitylated sites in maize seedling leaves
In order to investigate the time-course response of the maize ubiquitylome to light, total proteins were extracted from four groups of etiolated B73 maize seedling leaves exposed to light for 0 h, 1 h, 6 h, or 12 h (Figure 1). The MS intensities of the ubiquitylated peptides in each sample were detected. After the protein samples were digested with trypsin to expose the lysine (K) residues, the samples were subjected to two sequential diGly immunoprecipitations. Two sets of proteomics data representing two biological replicates(Rp1 and Rp2,each containing two technical replicates) were acquired for each group(1 h, 6 h, and 12 h) of maize seedling leaves. For the etiolated leaf sample (0 h), data for only three replicates were acquired instead of four. Clear differences in the correlations between ubiquitylomes could be seen between the groups of leaves that had undergone light treatment for 0 h, 1 h, 6 h, and 12 h(Figure S1).

Figure 1 Workflow for the time course proteomic analysis of the ubiquitylome in de-etiolating maize seedling leavesDark-grown B73 maize seedlings with ~7 days old (0 h) were exposed to light for 1 h,6 h,or 12 h before protein extraction from leaves and protein digestion by trypsin. Two sequential diGly immunoprecipitations were carried out to isolate the peptides containing K-ε-GG remnants that resulted from trypsin digestion of proteins with ubiquitylated lysines. Identification of these peptides was achieved by detecting a 114.04 Da adduct on the modified lysine residues using high-resolution MS (LTQ Orbitrap Velos). The relative quantitation of the ubiquitylated peptides in each sample was based on the MS intensities and was performed with the label-free quantitation model in MaxQuant(V1.3.5).K-ε-GG, di-glycine modified lysine; MS, mass spectrometry; LTQ,linear trap quadrupole.
In total, 1941 sites corresponding to 1053 proteins were identified (Data S1). It is important to note that 14 ubiquitylated sites (Ub sites) could be simultaneously assigned to multiple protein orthologs, of which 13 sites corresponded to 26 proteins and a peptide corresponded to 3 proteins. In total,15 redundant proteins were produced. Eliminating these repeatedly assigned Ub sites, a total of 1926 unique Ub sites were identified in our study.These Ub proteins contained various numbers of Ub sites, ranging from 1 to 19 per protein(Table 1), reflecting variation in the amount of ubiquitylation between proteins. Proteins containing the highest number of Ub sites included CDC48 (GRMZM2G036765_P01), sucrose synthase 2 (SUS 2)(GRMZM2G152908_P01), and a few heat shock protein (HSP) family members (each containing 10-11 Ub sites).In summary,we obtained the first large-scale proteome with time-course quantification in maize, and we obtained the largest coverage of the ubiquitylome of a C4 plant.

Table 1 Distribution of Ub sites in all identified Ub proteins
Consensus motif of ubiquitylation sites
To identify the specific motifs for ubiquitin attachment in the substrates, we searched for potential consensus ubiquitylation motifs in the identified Ub sites using the online tools motif-x(http://motif-x.med.harvard.edu/motif-x.html) [25,26] and ice-Logo (http://iomics.ugent.be/icelogoserver/create) [27]. Only the unique Ub sites identified in this study that were located more than five residues from the C terminus of the protein(1880 sites)were used for motif-x analysis.Five ubiquitylation motifs, namely KA, AXK, KXG, AK, and TK (amino acid residues are indicated by single-letter abbreviations, where X represents any amino acid residue) were identified, and these accounted for 838 (45% ) of the Ub sites (Figure 2). This is one of the few successes in the many attempts to detect ubiquitylation motifs[17,28-30].Guo et al identified EK,EXXXK,KD,KE,and KXXE as putative ubiquitylation motifs in petunias [30]; Xie et al identified seven putative ubiquitylation motifs, AAXXXXK, AXXXXXKXA, AXXXK, AK, EK,KXXA, and AXK, in rice [17]; and Zhang et al identified KXA,KXXA,AXXXXK,and TXK in wheat[18].The motifs EXXXK, AAXXXXK, AXXXXXKXA, AXXXK, AK,KXXA,AXK,KXA,KXXA,AXXXXK,and TXK identified in these species were also frequently found in our maize data,while the others were not (Figure 2).
We next generated a heatmap representing the -6 to +6 residues in the vicinity of the ubiquitinated lysine (ubK) using all 1926 unique Ub sites to investigate the residues in the ubiquitylation motif. We observed significant enrichment of alanine both upstream and downstream of the ubK(Figure 2C). Enrichment of lysine was observed downstream of the ubK, but depletion was observed upstream. Threonine and serine were enriched from -2 to +1. Glycine, valine,and glutamine were enriched at positions -1, +2, and +3.Notably, 46 (2.4% ) of the 1926 unique Ub sites identified in this study were located close to (within 6 residues) the Cterminus of the protein, but no Ub site located close to the N-terminus was identified. In summary, the ubK is generally surrounded by residues with small, polar, uncharged,hydrophobic, or basic side chains, similar to what was observed in rice and wheat[17,18].General enrichment of alanine and depletion of histidine and tryptophan are consistent with observations in maize and petunia[15-17,30].These findings suggest that ubiquitylation motifs in higher plant species share common features.
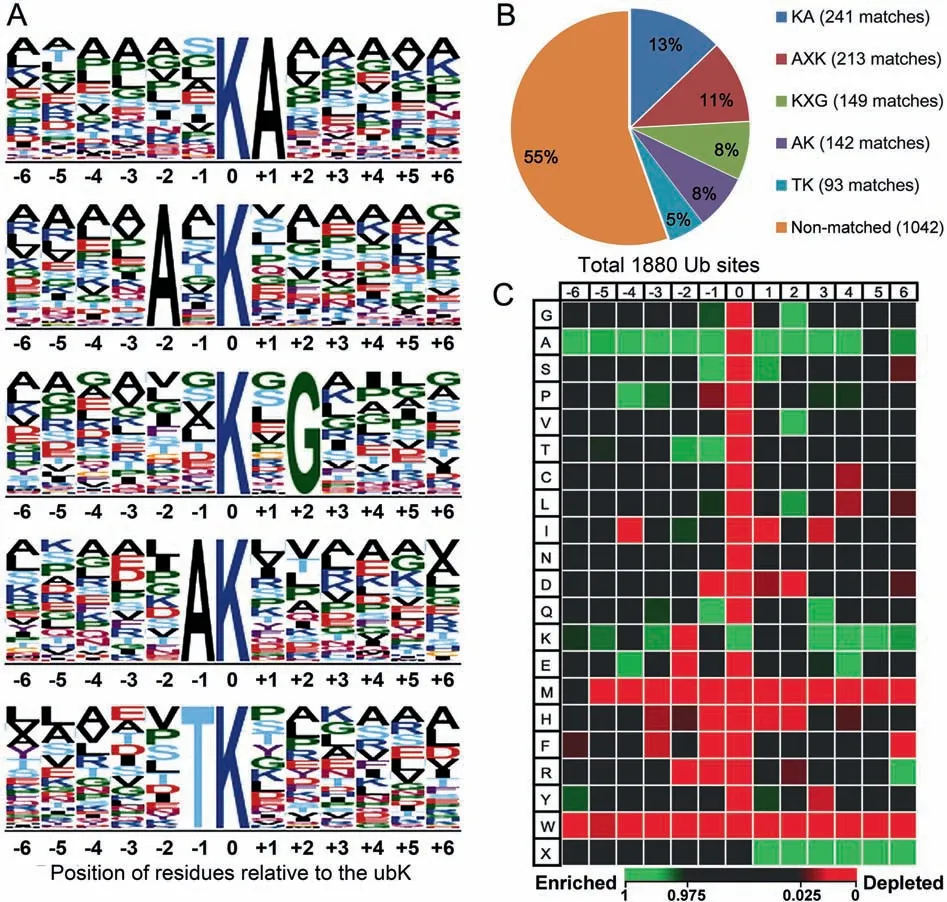
Figure 2 Ub site motif analysisConsensus motifs surrounding K at 1926 unique Ub sites were identified using motif-x and iceLogo.A.Motifs identified using motif-x with the dataset UniProt: UP000007305 as the background. B.Number of sequence matches for each potential motifidentified using motif-x.Five potential motifs identified using motif-x accounted for 838 (45% ) of the 1880 Ub sites located more than five residues from the C terminus of the protein.C.Heatmap representation of the-6 to+6 residues surrounding the ubK generated by iceLogo using all 1926 Ub sites.The precompiled Swiss-Prot composition of Zea mays L.was used as the reference(P <0.05).Enrichment and depletion of an amino acid residue relative to the random expectation were indicated in green and red,respectively.ubK,the ubiquitylated lysine.X,any residue.
Apart from the similarities mentioned above, the motifs were also quite different from previous study in plant.Methionine is enriched at +1 and +2 in Arabidopsis but is severely depleted in maize. The significant depletion of acidic residues near the ubK in maize (within ±2 residues) is opposite to what was observed in rice and petunia.In rice,the motif EK accounts for the highest number of Ub sites (~120 Ub sites, ~22% of the total) [17]. In petunia, both aspartic acid and glutamic acid are significantly enriched at multiple positions, and no depletion is observed at residues located within the -10 to +10 positions. Similarly, in Arabidopsis, the -1 to-3 and +3 positions are enriched in glutamic acid [15-17].
It appears that there are common and distinct features of ubiquitylation motifs between humans, yeast, and plants. Ub motifs in humans show enrichment of acidic residues and depletion of basic residues surrounding the ubK (-6 to +6 positions),as does petunia[31].Nevertheless,our findings generally suggest that Ub motifs in maize are distinct from those in humans and petunia. Nonetheless, another study of the human ubiquitylome found no significant sequence recognition motif, but detected enrichment of positively charged residues (lysine and arginine) upstream of the ubK [32]. This is also different from what we observed in maize: significant depletion of arginine upstream of the ubK (especially at the-1 and -2 positions). In addition, exclusion of downstream cysteine and the enrichment of alanine and glycine in the region surrounding the ubK were also reported in humans[32], which also agrees with our findings in maize. An earlier yeast ubiquitylome study also reported exclusion of cysteine surrounding the ubK. This is consistent with our finding in maize and the previous finding in petunias [33]. Based on our findings and previous reports from various species,variation exists in the preference of lysine for ubiquitylation.The inconsistencies between studies might be due to the species-specificity of ubiquitylation motifs. Another possible reason is biases in the types of motifs recovered from different purification methods [16,34].

Figure 3 GO enrichment analyses of Ub proteins in de-etiolating maize seedling leavesPlant GO slim terms that were found to be significantly overrepresented in Ub proteins (q value ≤0.05) using the topGO R language package are sorted by biological process(A),cellular component(B),and molecular function(C).Protein ratio indicates the proportion of Ub proteins among the total number of maize proteins assigned to each term. Ub, ubiquitylated.
Enrichment analyses of ubiquitylated substrates
To gain a global understanding of the functions and subcellular distributions of the identified ubiquitylation targets, Gene Ontology (GO) enrichment analyses were performed using 1041 Ub proteins with annotations in the Ensembl Zea_mays AGPv3.25 database (Figure 3). The enrichment of the cellular component (CC) category plastid indicates the involvement of ubiquitylation in the formation of chloroplasts [35]. The enrichment of the biological process (BP) category translation, the molecular function (MF) category translation factor activity, as well as the CC category ribosome suggests that ubiquitylation might be an important post translational modification (PTM) regulating protein synthesis. Additionally, 19 proteins involved in G-protein and receptor kinase signaling and 32 kinases/phosphatases/phosphodiesterases were annotated to the MF term nucleotide binding. These findings implicate crosstalk between ubiquitylation and phosphorylation of signaling proteins during de-etiolation of the maize leaf.
To investigate the general physiological significance of ubiquitylation throughout the 12-h de-etiolation period,Map-Man functional category enrichment analysis was carried out using all identified Ub proteins (Figure 4). The large number of Ub proteins in the protein degradation and protein synthesis is consistent with the known regulatory role of ubiquitylation in protein turnover. Both the number of Ub proteins and ratio of Ub proteins to all maize proteins in the functional protein synthesis are larger than those for protein degradation.This is presumably related to the rapid accumulation of photosynthetic proteins in the leaf during de-etiolation. The enrichment of photomorphogenesis-related proteins, which include the proteins ABCB19, ZmOrphan117, brachytic2, phytochrome A1, phytochrome A2, and CSN2, also adds to the evidence that ubiquitylation plays a regulatory role in photomorphogenesis [16].
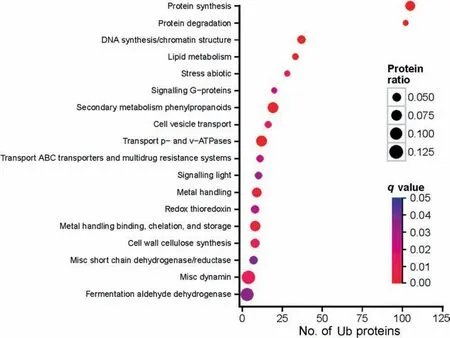
Figure 4 MapMan functional category enrichment analyses of the Ub proteins in de-etiolating maize seedling leavesThe enriched MapMan functional categories were analyzed using the topGO R language package. Protein ratio indicates the proportion of Ub proteins identified in this study among the total number of maize proteins in each pathway.
Interaction network of ubiquitylated proteins in de-etiolating leaves
The interactions between all identified Ub proteins were determined using STRING 10 [36], and a network (undigraph) with 638 nodes (Ub proteins) and 7729 edges (interactions) was generated(Figure 5A-C). In the visualized network image, each node represents an Ub protein, and the degree represents the number of interactions (Data S2). Based on our statistical analyses, the node degree for ribosomal Ub proteins was 102 on average, which is approximately twofold higher than the average node degree of all other Ub proteins in the network. Five other functional categories had a higher degree than average, indicating more interactions than average. Dense interactions were evident for ubiquitinated proteins annotated to the categories (1) protein synthesis,(2) glycolysis cytosolic branch, (3) DNA synthesis/chromatin structure, (4) protein degradation, (5) stress abiotic, as well as (6) transport p- and v-ATPases, indicating that all of these categories include proteins involved in the ubiquitin-proteasome pathway.
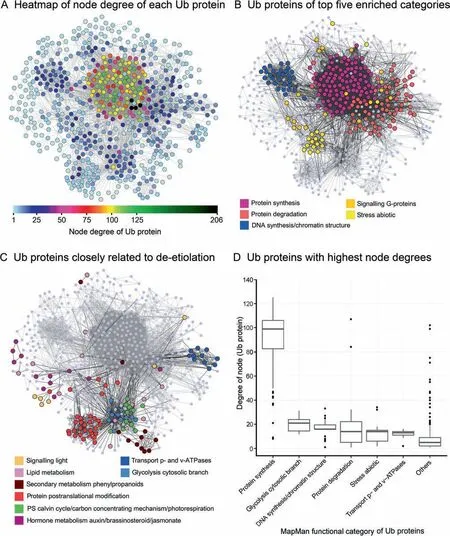
Figure 5 Interaction networks for all identified Ub proteins in the ubiquitylome of de-etiolating maize seedling leavesThe interaction networks for all Ub proteins identified in this study were generated using STRING 10(minimum required interaction score≥0.7) with all active prediction methods and visualized using Cytoscape software (Ver. 3.2.1). The prefuse force-directed layout with manual adjustments was used to uncover overlapping nodes and to fit the network into a compact scale for display. Proteins (nodes) are shown as bubbles filled with different colors;bubble size and border width indicate functional categories.The related interactions(edges)for the selected proteins are shown as black lines, and interactions (edges) between other proteins are shown as gray lines. A. A heatmap representing the node degree of each protein in the interaction network of all identified Ub proteins. The range of degree distribution is shown as a color gradient from light blue to dark green. A significant central interaction region and multiple peripheral hubs can be observed.B.Ub proteins belonging to the top five enriched MapMan categories.Different categories are indicated by the node fill color.C.Ub proteins classified into functional categories that are closely associated with the de-etiolation process.Different categories are indicated by the node fill color. D. Box plot showing the node degree of the functional categories containing Ub proteins with the highest average node degrees. ‘‘Others” indicates proteins in categories other than the six indicated categories. The functional categories are displayed along the Y axis in order of descending average node degree. The node degrees originally ranged from 1 to 206; here only nodes with degrees ranging from 0 to 125 are shown. See Figure S2 for the original box plot.
The node degrees for the Ub proteins in each of the six functional categories above were analyzed (Figure 5D, Figure S2). Wilcoxon Rank Sum test showed that the six categories indeed shared significantly more interactions than the‘‘others” (P <0.01). According to the Kruskal-Wallis test,there were significant differences in node degree between the six functional categories and ‘‘others” (P <0.01), but no significant difference was found between the five categories except for ‘‘protein synthesis” (P >0.05). These results indicate that in de-etiolating maize leaves, ubiquitylation plays an important role in the following processes: cytosolic glycolysis,DNA synthesis/chromatin structure regulation,protein degradation, response to abiotic stress, and transport p- and v-ATPases.In particular,ubiquitylation has a strong influence on protein synthesis.Compared with these processes,ubiquitylation may play a more important role in the six processes listed above than in other physiological processes.
In the ubiquitylation network, three of the four proteins with the highest node degrees (205-206) were polyubiquitin proteins (one is an unknown protein). Four 40S ribosomal protein S27a (RPS27a or homologs) proteins also had high node degree (107) (Data S3). Polyubiquitin and natural ribosomal-ubiquitin fusion proteins such as ubiquitylated RPS27a are the two sources of monoubiquitin in the cell[37,38]. This might explain why the polyubiquitin proteins have high node degrees.
Relative quantification of ubiquitylation substrates
Among the 1926 unique Ub sites identified, 654 sites corresponding to 459 Ub proteins were identified at all time points in all our replicates (Data S4). The time-course fold change(FC) in the MS intensity of peptides containing these 654 Ub sites relative to the time point 0 h, namely 1 h/0h (FC1),6 h/0h (FC6), and 12 h/0h (FC12), were calculated. As in previous publications [39,40], we defined up/down-regulation based on an FC threshold value of 1.5 and 0.7 for up- and down-regulation, respectively (Table S1). Stringent criteria were established for defining the FC in the Ub sites over the time course (Table S1). When comparing Rp1 and Rp2, 214 reproducibly quantified Ub (qUb) sites corresponding to 173 proteins, namely qUb proteins, showed consistent regulation(Data S5).
Seventy-eight (36% ) of the qUb sites showed time-course regulation according to our FC thresholds as described in Table S1. The ubiquitylation level was upregulated at most of these sites (Up, 22% of all qUb sites). Hierarchical clustering analyses were performed using the FC results for two clusters of qUb sites: all qUb sites (Figure S3A), and time-courseregulated qUb sites (Figure S3B). The clustering results indicate that there was comparable up-regulation of ubiquitylation at 1 h and at 12 h, suggesting that some ubiquitylation events take place at a very early stage of de-etiolation (1 h). These ubiquitylation events possibly correspond to the early response to light. The ubiquitylation levels of some sites gradually increased and reached peak levels at the 12 h time point.
During the 12-h illumination, the 78 regulated qUb sites showed altered ubiquitylation levels, compared with those at 0 h, with ratios ranging widely from 0.3 fold (K172 in sucrose synthase 2, GRMZM2G152908_P01) to 29.9 fold (K301 in a maize homolog of Arabidopsis allene oxide synthase (AOS),GRMZM2G067225_P01) (Table 2). The remaining 136 qUb sites showed no significant change in ubiquitylation(NS)(Figure 6A). Of these proteins, 15 (9% of all qUb proteins) contained qUb sites that were differentially regulated(Figure 6B). The percentage of qUb proteins that were regulated (40% , 69 out of 173) was only slightly higher than the percentage of regulated qUb sites (Figure 6C). Notably, 26(15% ) of the 173 qUb proteins contained multiple qUb sites.
To investigate the correlation between the changes in ubiquitylation levels and protein levels, we used the 172 qUb proteins with annotations in MaizeGDB as queries to search our protein quantification data (ProteomeXchange: PXD012897)that were acquired using nanoLC MS/MS. The leaf samples used for this analysis were taken from maize seedlings that underwent the same treatments as those used for analysis of ubiquitylation. There were 86 protein hits (Data S6) for 109 qUb sites and 35 protein hits for 40 regulated qUb sites(Data S5).Surprisingly,only six of all qUb site hits,including five of the regulated qUb site hits, showed time-course alterations in protein level.These results suggest that the level of ubiquitylation,but not protein abundance,changed for most of the qUb proteins uncovered in this study. Deubiquitinating enzymes(DUBs) play a counteracting role against ubiquitylation [41]and are known to have multiple regulatory functions in plants[42]. Thus, the time-course regulation of qUb sites in maize is possibly the consequence of ongoing antagonism between ubiquitylation and deubiquitylation.

Table 2 qUb sites with time-course alterations in ubiquitylation level

Table 2 qUb sites with time-course alterations in ubiquitylation level
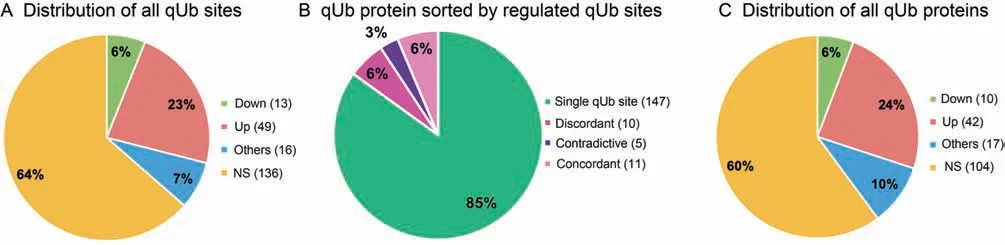
Figure 6 Distribution of qUb sites and qUb proteins according to time-course alteration in ubiquitylation levelPercentages and numbers of qUb sites/proteins for each of the categories,NS,Up,Down,and Others,are shown.See Table S1 for more details.A.All qUb sites.B.qUb proteins sorted according to the presence of single or multiple qUb sites.qUb proteins with multiple qUb sites are further classified as discordant, contradictive, or concordant based on the pattern of time-course alteration in ubiquitylation levels.Discordant,qUb protein containing both an NS Ub site(s)and a qUb site(s)from only one of the three categories Up,Down,and Others; contradictive, a qUb protein containing qUb sites from at least two of the three categories Up, Down, or others; concordant, a qUb protein only containing qUb sites from one category. C. All qUb proteins sorted according to their time-course alteration in ubiquitylation level as defined in Table S1. qUb site/protein, ubiquitylated site/protein quantified with repeatability; NS, no significant change; Up, up-regulated, Down, down-regulated; Others, pattern of change in ubiquitylation other than NS, Up, or Down.
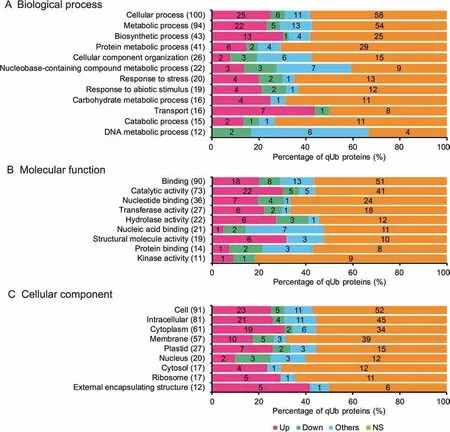
Figure 7 Distribution of qUb proteins among GO termsThe total number of qUb proteins assigned to each GO slim term containing >10 Ub proteins is shown;see Figure S4 for more detailed GO terms. The percentage and number of qUb proteins assigned to each group Up, Down, Others, and NS (according to Table S1) are shown on the horizontal axis and to the right of the GO category name, respectively. A. Biological process. B. Molecular function. C.Cellular component.
Enrichment analyses of time-course-regulated ubiquitylated proteins
The percentage of qUb proteins annotated to each GO term and MapMan category with changes in ubiquitylation level(according to the thresholds 1.5/0.7) were visualized using bar graphs (Figure 7, Figure 8, Figures S4 and S5). The qUb proteins involved in DNA metabolism had highly varied changes in ubiquitylation levels. These proteins were annotated to the GO BP terms metabolic process, nucleobasecontaining compound metabolic process, and cellular component organization, the MF term nucleic acid binding, and the MapMan category DNA synthesis/chromatin structure.Such regulations mainly reflect diverse and complex histone ubiquitylation and the changes in the ubiquitylation levels of these proteins;60% -100% of the qUb proteins in each of these categories were histone components (Data S5 and S8). The number of regulated qUb proteins (67 in total) annotated to each GO term or MapMan category was also visualized in bar graphs (Figure 9). This analysis suggests that the proteins with regulated qUb sites are primarily involved in protein and DNA metabolism in the de-etiolating maize leaf (Figure 9D).
A high percentage of the regulated qUb proteins involved in protein metabolism were up-regulated. These proteins were annotated to the MF term structural molecule activity,the CC term external encapsulating structure, and the MapMan category protein synthesis, and mainly consist of ribosomal proteins (100% , 67% , and 70% of the proteins in the MF, CC,and MapMan categories, respectively) (Figures 7 and 8, Data S5 and S8). Notably, a higher percentage of regulated qUb proteins were annotated to the MapMan category protein synthesis than to protein degradation.
A high percentage of regulated qUb proteins(>50% )were also annotated to the MapMan categories jasmonate metabolism, C4 carbon concentrating mechanism, ABC transporters,and major CHO metabolism (Figure 8). This indicates the active regulatory role of ubiquitylation in these processes during de-etiolation.The regulated qUb proteins annotated to the BP term transport were mostly(88% )up-regulated(Figure 7).These include one non-specific lipid-transfer protein(GRMZM2G101958_P01), one carbohydrate transporter(GRMZM2G029219_P01),and two vacuolar ATPase subunits(Data S5).
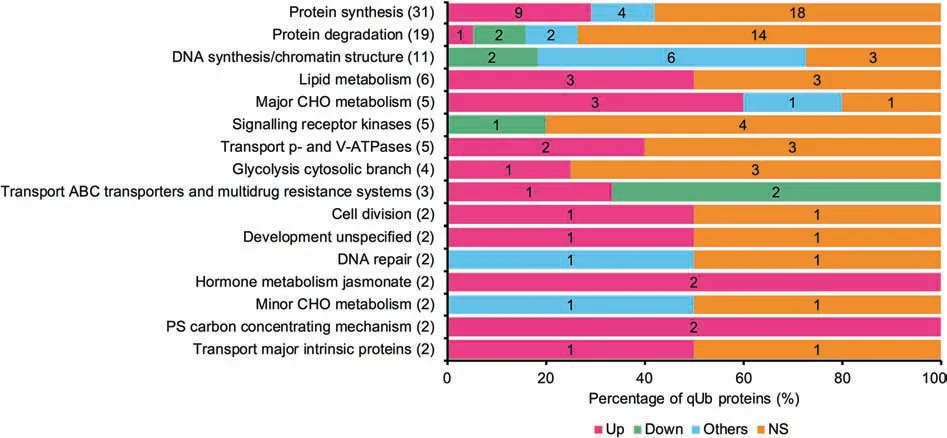
Figure 8 Distribution of qUb proteins among MapMan functional categoriesThis figure only shows MapMan categories with >1 qUb with time-course alteration in ubiquitylation level; see Figure S5 for all MapMan categories.The total number of qUb proteins assigned to each MapMan functional category is shown on the vertical axis.The percentage and number of qUb proteins assigned to each group Up, Down, Others, and NS (according to Table S1) are shown on the horizontal axis and to the right of the MapMan category name, respectively.
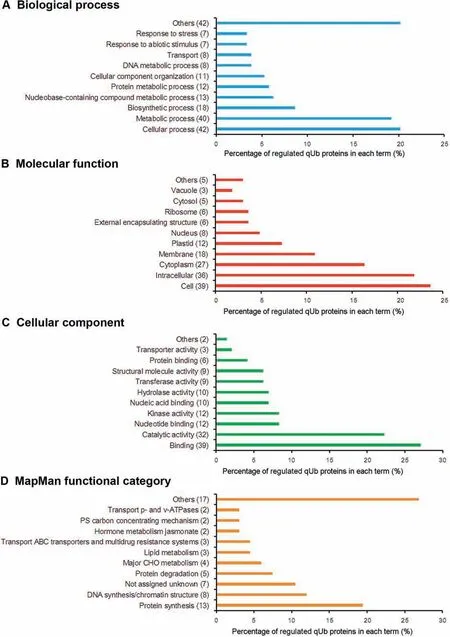
Figure 9 Distribution of regulated qUb proteins among the top 10 most abundant GO slim terms/MapMan functional categoriesThe distribution of all time-course regulated qUb proteins among the top 10 terms/categories with the highest number of qUb proteins is shown.The percentage of the regulated proteins in each term/category is indicated on the bar graph,and the number of proteins is noted in the legend. A. GO biological process. B. GO molecular function. C. GO cellular component; D. MapMan functional categories.
We looked at the locations of these regulated qUb proteins in the interaction network (Figure 5) to gain a general understanding of their roles in de-etiolation(Figure S6).An interaction network of the regulated qUb proteins was also generated(Figure 10). The layout and composition of the qUb network(Figure 10)are in general consistent with those of the network generated using all Ub proteins(Figure 5A-C).Both networks have a large central region with dense interactions.The central region of the qUb network is mainly composed of qUb proteins involved in protein synthesis (30) and protein degradation (9). The qUb proteins annotated to a highly enriched MapMan category (Figure 8), DNA synthesis/chromatin structure, form a small independent sub-network.
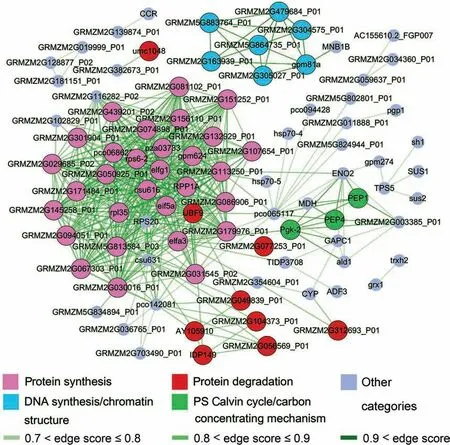
Figure 10 Interaction networks for all qUb proteins in the de-etiolating maize seedling leaf ubiquitylomeAn interaction network of the qUb proteins was generated using STRING with the same score setting used for all identified Ub proteins(Figure 5).The top three enriched MapMan functional categories and photosynthetic qUb proteins are indicated using different fill colors.The combined scores of the interactions and edge scores provided by STRING(Data S8)are shown in green with different intensities from light to dark indicating edge scores of 0.7-0.8, 0.8-0.9, and >0.9.
According to the results of our enrichment and proteinprotein interaction analyses, we took a closer look at substrates involved in protein degradation, protein synthesis,and histone modification. In addition, since the changes in photosynthesis, light signaling, hormone metabolism, and sink-source transition are key events during de-etiolation, we focused on the ubiquitylation substrates annotated to these categories, and discuss these substrates in the following sections.
Protein ubiquitylation is regulated in de-etiolating maize seedling leaves
We found 128 Ub sites corresponding to 82 Ub proteins in the ubiquitin degradation pathway, and five sites were regulated.Three Ub sites in two maize homologs of Arabidopsis regulatory particle AAA-ATPase 2a (RPT2a; K63 on GRMZM2G104373 and K64 and K245 on GRMZM2G056569) were identified/quantified. Site K63 in GRMZM2G104373_P01 showed stable down-regulation throughout the 12-hour de-etiolation despite there being no change in protein level (Data S1, S4, and S5).
RPT2 is a base component of the 19S regulatory subunit in the 26S proteasome [43]. RPT2a, one of the two homologous genes that encode RPT2 in Arabidopsis, has been reported to influence various developmental processes in plants, including maintenance of root/shoot apical meristem activities and regulation of leaf organ size and cell proliferation[44-46].Previous reports have suggested that in various eukaryotes,proteasome subunits are subjected to PTMs, both reversible and irreversible. Such modifications include the formation of protein carbonyls, the addition of N-acetylglucosamine, Sglutathionylation of cysteineresidues, acetylation, phosphorylation, and possibly ubiquitylation. These modifications can dynamically alter the subunit structure and thus proteasomal activity [47]. However, there have been no reports of sitespecific ubiquitylation of RPT2a. The finding that maize RPT2a is ubiquitinylated suggests that ubiquitylation might be an important PTM regulating this protein. The change in ubiquitylation level during de-etiolation suggests that RPT2a might have a regulatory role in photomorphogenesis. In addition, ubiquitylation might be an important PTM regulating this protein.
Ubiquitylation at K377 of maize copine-1(GRMZM2G345544), which shares high sequence similarity with RING DOMAIN LIGASE2 (RGLG2) in Arabidopsis,showed sustained down-regulation after 1 h of light treatment.Copine is a class of highly conserved proteins present in almost all organisms[48].However,there have been few reports on its physiological functions. In Arabidopsis a copine gene, CPN1,regulates responses to low humidity [49] and in Mesembryanthemum crystallinum, CPN1 interacts with suppressor of K+transport growth defect 1 (McSKD1) and regulates the maintenance of Na+/K+homeostasis under salt stress [50].RGLG2 modulates the directional flow of auxin through the formation of Lys-63-linked polyubiquitin chains in Arabidopsis[51]. These reports suggest that copine proteins are likely involved in plant responses to abiotic stimuli.
Ubiquitylation of ribosomal and histone proteins is regulated in de-etiolating maize seedlings
Multiple ribosomal Ub proteins have been identified in previous proteomic studies in Arabidopsis and other species[16,52,53].In this study,we identified 175 Ub sites corresponding to 76 ubiquitylated ribosomal components,and the regulation of 23 qUb sites in 21 ribosomal qUb proteins was determined.Nine of these sites showed time-course alterations in ubiquitylation levels(Data S1,S5,and S6).These sites include K138 in 40S ribosomal protein S12(GRMZM2G132929,~28 fold at 12 h),K144 in a maize homolog of 40S ribosomal protein S10 (RPS10C)(GRMZM2G050925_P01,~8 fold at 12 h),and K145 in ribosomal protein S10 (GRMZM2G171484_P01, ~7 fold at 12 h)(Data S4,S5).
Ubiquitin-40S ribosomal protein S27a(GRMZM2G431821, RS27a) was drastically up-regulated(~26-fold increase at 12 h) at K63, despite being stably expressed throughout the 0 h to 12 h de-etiolation process(Data S1, S5, and S6). In total, 11 Ub sites were identified in RS27a. The Ub sites K6, K11, K27, K29, K33, K48, and K63 were detected in the N-terminal ubiquitin part of this fusion protein. Ub sites in the N-terminal regions of proteins are where poly-ubiquitin chains with various topologies are usually formed. Ub sites in the C-terminal region of RS27a(sites K107,K114,K144,and K154)were also identified(Data S1, S5, and S6). Ubiquitin-ribosomal fusion proteins are the natural products of ubiquitin-encoding genes and are further processed by specific enzymes into monoubiquitin and ribosomal subunits[37].In mammals,ribosomal proteins encoded by these genes have been shown to have extraribosomal functions[54,55].Expression of RS27 was reported to vary between callus and early-stage development in Cyclamen persicum [56],which might indicate an extraribosomal function in plants.
Ubiquitylation is a well acknowledged and important histone PTM [57]. Some of the previous ubiquitylome studies in higher plants failed to detect a large number of histone substrates[16].However,in our study,77 Ub sites in histone components were identified,and 11 qUb proteins with 16 qUb sites were quantified (Data S1, S5, and S6). The monoubiquitylation of H2A and H2B has been recognized as an important epigenetic mark in higher plants [58]. In our study, multiple Ub sites on H2A and H2B, as well as in H1, H3.2, and H4 were identified/quantified. The regulation of ubiquitylation of histone proteins during maize leaf de-etiolation is complex. For example,in maize H2B(GRMZM2G304575_P02),the ubiquitylation level of K134 was slightly up-regulated throughout deetiolation, whereas the ubiquitylation of K105 was downregulated after 1 h, and K111 showed varying degrees of upand down-regulation.
Ubiquitylation of key C4 photosynthetic enzymes and sink-tosource transition-associated proteins is regulated in de-etiolating maize seedlings
Seven Ub sites in maize phosphoenolpyruvate carboxylase 1(PEPC 1, GRMZM2G083841_P01), and three Ub sites in other PEPC isoforms, e.g., maize PEPC isoform 1(GRMZM2G074122_P02), as well as in maize homologs of the Arabidopsis PEPCs ATPPC1 (GRMZM2G473001_P01)and ATPPC3(GRMZM2G110714_P02)were identified/quantified (Data S1, S5, and S6). K734-ubiquitylation of maize PEPC 1 was up-regulated significantly (~8.5 fold at 12 h),despite only a slight increase in the protein level (~2.2 fold at 2 h). These results indicate that the proportion of ubiquitylated PEPC 1 protein increased during maize de-etiolation.The monoubiquitylation of PEPC at K628 in castor bean and K624 in sorghum, a strictly conserved residue, has been shown to regulate the catalytic activity of PEPC[59,60].Interestingly, the corresponding homologous sites in maize were both identified in the present study: K624 of maize PEPC isoform 1 (GRMZM2G074122) and K634 of maize PEPC 1(GRMZM2G083841)(Data S1).Our data show that ubiquitylation at all these sites was up-regulated during de-etiolation(>3 fold), except for a few time points in some experimental replicates where quantification was not achieved (Data S1).
Intriguing ubiquitylation profiles were also found for maize carbonic anhydrase (CA). Two Ub sites in GRMZM2G348512_P06 and seven Ub sites in GRMZM2G121878_P02 were identified. These sites were not ubiquitylated(MS intensity of 0)at 0 h and 1 h but ubiquitylation was detected at 6 h and 12 h. Thus, the ubiquitylation of these maize CA sites is potentially light induced (Data S1,Table S2).This regulatory pattern suggests that the ubiquitylation of these sites might be reversible and might serve as a mechanism for fine-tuning the catalytic activity of CAs. Thus,ubiquitylation might directly regulate de-etiolation by altering the activities of key photosynthetic enzymes.Enzymes involved in the Calvin cycle were also found to be ubiquitylated.Six Ub sites in phosphoglycerate kinase (GRMZM2G382914_P03),two Ub sites in fructose-bisphosphate aldolase(GRMZM2G046284_P02), and one Ub site in NADPdependent glyceraldehyde-3-phosphate dehydrogenase(GRMZM2G035268_P03)were identified.Two Ub sites in photosystem II components and one Ub site in a photosystem I component,K10,which is located in the chloroplastic photosystem I reaction center subunit VI,were also identified.
In addition, ubiquitylation of light signaling proteins was also identified. Three Ub sites in COP9 signalosome subunit one(GRMZM2G062210_P01),two Ub sites in a maize homolog of Arabidopsis phytochrome B (GRMZM2G124532_P03),and 11 Ub sites in phytochrome A homologs (two Ub sites in GRMZM2G157727_P02, nine in GRMZM2G181028_P01)were identified (Data S1). Five qUb sites in GRMZM2G181028_P01 showed light-induced downregulation of ubiquitylation. Ubiquitylation at these sites was detected and quantified in all replicates at the 0 h and 1 h time points,but was not detected at the 6 h and 12 h time points.This decrease in ubiquitylation coincided with a drastic drop in protein level(Data S1,S6,and Table S3),presumably reflecting the rapid degradation of phytochrome A via the ubiquitin-proteasome system during exposure to light.
In this study, 22 Ub sites in four maize sucrose synthases(SUS) and one Ub site in a sugar transporter were identified/quantified(Data S1,S6).The regulation of SUS protein level is important during the sink-to-source transition in plant organs[61].Thisprocessisalsocorrelatedwiththeaccumulationofphotosynthetic enzymes and sucrose biosynthetic enzymes such as sucrose-phosphate synthase [61]. Although we did not detect degradation of SUS within the first 12 h of de-etiolation, we did identify ubiquitylation on SUS proteins.We detected significant down-regulation of ubiquitylation at K172(0.3 fold at 6 h)and slight up-regulation (1.8 fold) at K327 in SUS 2(GRMZM2G152908_P01,maize homolog of Arabidopsis SUS 4)only at 12 h.Sustained up-regulation (2.3-3.5 fold from 1 h to 12 h) in ubiquitylation at K539 in SUS 3 (GRMZM2G 318780_P02)was observed.In addition,up-regulation of K170 ubiquitylation in the sugar transporter (GRMZM2G0292 19_P01)was observed(Table 2,Data S5).Together,these findings imply that ubiquitylation might play a regulatory role in the sink-to-source transition during de-etiolation.
Ubiquitylation of hormone, lipid and secondary metabolism proteins is regulated in de-etiolating maize seedlings
Ubiquitin-proteasome-directed degradation is a key regulatory mechanism in the signaling pathways of multiple hormones including auxin, jasmonate, gibberellin, brassinosteroid, and ethylene[62].We identified/quantified 59 Ub sites representing 31 proteins involved in hormone metabolism/signaling/trans port(Data S7).Multiple Ub sites,some exhibiting altered ubiquitylation levels, in hormone biosynthetic enzymes were also found (Data S7). For example, a maize homolog(GRMZM2G067225_P01) of Arabidopsis AOS, an enzyme involved in jasmonate synthesis [63], showed drastic upregulation of ubiquitylation at both K301 (up to ~30 fold)and K320(up to ~11 fold)despite there being no change in protein level (Data S5). Thus, ubiquitylation might regulate deetiolation by directly influencing plant hormonal homeostasis.
We identified/quantified 68 Ub sites representing 33 proteins annotated to the category lipid metabolism. These include 21 Ub sites in five maize 3-ketoacyl-CoA synthases:two Ub sites in GRMZM2G003501_P01,five in GRMZM2G032095_P01,two in GRMZM2G164974_P01, two in GRMZM2G168304_P01,and ten in GRMZM2G445602_P01(Data S1,S4).Ubiquitylation at K433 in GRMZM2G445602_P01 showed sustained upregulation(up to ~3.6 fold at 12 h).Ubiquitylation at K141 in GRMZM2G445602_P01 was moderately up-regulated at 6 h and 12 h (2.3 fold at 6 h and 2.0 fold at 12 h) (Data S4). Ten Ub sites in five enzymes for phospholipid synthesis were identified/quantified (Data S1, S4). Ubiquitylation at K159 in glycerol-3-phosphate acyltransferase 8 (GRMZM2G 059637_P01) was slightly up-regulated throughout de-etiolation (1.9-2.2 fold from 1 h to 12 h, Data S4).3-ketoacyl-CoA synthase is the initial enzyme for the production of very-long-chain fatty acids, which are essential precursors of cuticular waxes, aliphatic suberins, and membrane lipids including sphingolipids and phospholipids [64]. Thus,these substrates might be associated with leaf strengthening,plastid development, and membrane system activity during de-etiolation. Hence, our results suggest ubiquitylation plays regulatory roles in these physiological processes.
We identified/quantified 59 Ub sites corresponding to 31 proteins involved in secondary metabolism pathways including phenylpropanoid, flavonoid, isoprenoid, and wax metabolism(Data S1, S5). The ubiquitylation of a maize homolog(GRMZM2G139874_P01) of Arabidopsis trans-cinnamate 4-monooxygenase(ATC4H),an enzyme that catalyzes the second initial reaction of the phenylpropanoid pathway [65], was upregulated at K141 and K205(up to ~3.5 fold at 12 h).A maize homolog (GRMZM2G131205_P02) of Arabidopsis cinnamoyl-CoA reductase(CCR1),a key enzyme for lignification [66], showed sustained up-regulation of K134 ubiquitylation(up to ~5 fold)throughout the first 12 h of de-etiolation.
Since the cell membrane and the membranous structures of organelles such as vacuoles and plastids contain lipids,the identification/quantification of substrates for lipid metabolism(Figures 3-5) indicates the active involvement of ubiquitylation in membrane formation, transportation, and leaflignification.Thus, ubiquitylation presumably regulates chloroplast formation, nutrient uptake, and vascular development during deetiolation. A previous study found that ubiquitylation plays a role in driving the intracellular trafficking of a root iron transporter in Arabidopsis [67]. This provides evidence supporting our inference that ubiquitylation may regulate nutrient uptake.
Conclusion
Here we present the largest ubiquitylome dataset ever generated for a C4 plant, including site-specific and time-course quantification. We found that in the de-etiolating maize leaf,ubiquitylation may have important functions in the remodeling of both the proteome and the transcriptome. We identified/quantified a large number of Ub sites in substrates involved in G-protein, receptor kinase, and protein modification pathways (Figures 3-5, and Data S1). This provides evidence for PTM crosstalk and PTM superposition between ubiquitylation and phosphorylation. These PTMs might function together to fine-tune de-etiolation and potentially other transitions in the life cycle of plants.Our findings also revealed multiple Ub sites in key photosynthesis and light-signaling proteins and their distinct regulation,providing a basis for further investigation of whether and how the activities of these proteins are regulated by ubiquitylation.
We found that most of the regulated qUb proteins have altered ubiquitylation levels but unchanged protein levels.Previously we discovered that during the first 12 h of de-etiolation in maize leaves,the abundance of some proteins is maintained at a stable level while their transcripts rapidly accumulate (unpublished data).A possible explanation for this phenomenon is that the plant produces a stock of transcripts in response to acute environmental changes such as the dark-to-light transition.Thus,the plant prepares itself for making rapid and appropriate responses to altered growth conditions in the long term. This might be one of the mechanisms for adaptation in plants,which are immobile.Nevertheless,such preparations at the transcriptaccumulation level or protein-synthesis level are costly.In comparison,responses at the PTM level are more time-efficient and consume less energy. Given the reversibility of ubiquitylation regulated byDUBs,the site-specific ubiquitylation of a substrate might serve as a switch regulating alternative activities of proteins.Such regulation of site-specific ubiquitylation might have important roles during photomorphogenesis. Our findings enrich the knowledge about the functions of ubiquitylation in higher plants, and in particular provide insight into the roles of the ubiquitin system during C4 photomorphogenesis.
Materials and methods
Plant materials and sample collection
Maize (Zea mays L.) inbred line B73 was germinated in the dark and seedlings were cultured in pot soil with sufficient water in complete darkness at a constant temperature of 30°C for approximately 7 days until the first leaf had just fully expanded. Then the etiolated seedlings were divided into four groups with no fewer than 150 seedlings in each group. The four groups of maize seedlings were illuminated with white light(100 μmol m-2s-1)for different amounts of time,namely 0 h,1 h,6 h,and 12 h.After illumination,the first leaf of each seedling in each group(0 h,1 h,6 h,and 12 h)was swiftly harvested and wrapped in tinfoil. The collected leaf samples were immediately frozen in liquid nitrogen. A safe green light was used while watering and collecting samples in the dark.
Protein extraction
Protein extraction was performed as previously described with some modifications[12].Two biological replicate samples were separately ground in liquid nitrogen with a mortar and pestle sterilized by flaming. For each sample, 1.5 g of leaf powder was precipitated in precipitation solution[10% (w/v)trichloroacetic acid(TCA)and 0.07% (v/v)β-mercaptoethanol in acetone]at-20°C overnight.Thereafter,the sample was centrifuged at 40,000g for 1 h at 4°C,the supernatant was discarded,and the pellet was washed with rinsing solution [acetone containing 0.07% (v/v) β-mercaptoethanol] at -20°C and again centrifuged at 40,000g for 1 h at 4°C.The final protein pellet was vacuum-dried for 1-2 min and then solubilized in a pH 8.0 solution containing 6 M guanidine hydrochloride,50 mM Tris-HCl(pH 8.0), 40 mM DTT, and 1× protease inhibitor mixture(Roche,Basel,Switzerland)for 1 h at room temperature.Insoluble material was removed by centrifugation at 100,000g for 1 h at room temperature.Each protein sample was vacuum-dried to a powder and stored at-80°C for future processing.
Sample preparation
Each biological replicate from each group (0 h, 1 h, 6 h, and 12 h, ~5 mg protein powder each) was reconstituted with the addition of 300 μl water, and the pH was adjusted to >7.5 with 1 M Tris HCl (pH 8.1). Disulfide bonds were reduced by the addition of 20 mM DTT and incubation at 60°C for 30 min. After cooling to room temperature, 40 mM iodoacetamide was added and the samples were kept in the dark for 1 h to block free cysteine. The samples were dialyzed against 2 M urea and 50 mM NH4HCO3for 2 h and then twice against 50 mM NH4HCO3for 2 h each. Trypsin was added to the samples at a ratio of 1:50 (w/w, trypsin: sample), and the samples were incubated at 37°C overnight. The digested peptides were filtered using Amicon Ultra centrifugal filters(10 kD MWKO, Merck Millipore, MA) and dried under a vacuum.
Immunoaffinity purification
The ubiquitylation target proteins were isolated through immunoprecipitation using antibodies that recognize the Lysε-Gly-Gly (K-ε-diGly) remnants of trypsin-digested proteins with ubiquitylated lysines. The ubiquitylated peptides from each sample were enriched using the PTMScan Ubiquitin Remnant Motif (K-ε-GG) antibody (Catalog No. 5562, Cell Signaling Technology, Danvers) following the manufacturer’s instructions. The eluted peptides were desalted on an Empore 3M C18 (2215) StageTip1 (Sigma-Aldrich, Merck KGaA,Darmstadt, Germany) and digested again with 0.1 μg trypsin at 37°C for 2 h prior to nano LC-MS/MS analysis.
Nano LC-MS/MS analysis
Nano LC-MS/MS analysis was carried out primarily using a Dionex RSLC system interfaced with a Velos LTQ Orbitrap ETD (ThermoFisher, San Jose, CA). Samples were loaded onto a self-packed 100 μm×2 cm trap packed with Magic C18AQ (5 μm 200 A, Michrom Bioresources, Aubum, CA)and washed with Buffer A (0.2% formic acid) for 5 min at a flow rate of 10 μl/min. The trap was brought in-line with a homemade analytical column (Magic C18AQ, 3 μm 200 A,75 μm×50 cm) and peptides were fractionated at 300 nl/min with a multi-stepped gradient of 4% -15% Buffer B (0.16% formic acid, 80% acetonitrile) over 25 min, 15% -25% Buffer B over 65 min, and 25% -50% Buffer B over 55 min. MS data were acquired using a data-dependent acquisition procedure.A cyclic series of full scans were acquired in the Orbitrap with a resolution of 60,000,followed by MS/MS scans(38% of relative collision energy in the HCD cell) of the ten most intense ions. The settings were as follows: repeat count=one,dynamic exclusion duration=30 s, resolution for Orbitrap scanning=15,000, low mass=110 amu. In Nano LC-MS/MS experiments, the sample for each biological replicate for each group was loaded separately two times as two technical replicates. Thus, four sets of raw data were acquired for each group of samples.
Database searches and MS data analyses
The resulting MS/MS data were processed using MaxQuant with an integrated Andromeda search engine (version 1.5.2.8). Tandem mass spectra were searched against the Ensembl Zeamays AGPv3.25 database (containing 63,235 sequences). The peptides were also searched against a protein database translated from RNA-seq data from the same samples (ProteomeXchange: PXD012897, 14 proteins whose IDs start with ‘‘TCONS” are shown in Data S1). The protein descriptions were acquired from the UniProt and MapMan databases. For K-Ub peptides, trypsin/P was specified as a cleavage enzyme, and up to two missed cleavages were allowed. First, the search range was set to 20 ppm for precursor ions, and the main search range was set at 4.5 ppm and 0.5 Da for fragment ions. Carbamidomethyl on Cys was specified as a fixed modification, and GlyGly on Lys,deamidation on Asn or Gln, and oxidation on Met were specified as variable modifications. The false discovery rate (FDR) was adjusted to less than 1% at both the peptide and site levels,while the minimum score for modified peptides was set to greater than 40. In quantitative analyses, the intensities of enriched K-Ub peptides from the two biological replicates Rp1 and Rp2(each consisting of two technical replicates)were extracted from MaxQuant‘‘GlyGly(K)Sites.txt”.For the 0 h sample,the proteomic data for only three replicates,including one biological replicate and the two technical replicates of another biological replicate, were acquired (Figure S1). The 1 h/0h, 6 h/0h, and 12 h/0h ratios for each K-Ub peptide were calculated using the label-free intensities and normalized to the total intensity of all identified spectra.Spectra that had a ratio of ‘‘Divide 0” were replaced with an arbitrary number, ‘‘10”.
Quantification of ubiquitylation substrates
Relative quantitation of the ubiquitylated peptides in each sample was based on the MS intensity and was performed with a label-free quantitation model with a peptide FDR <1% at the protein,peptide,and site levels. Furthermore, we removed the peptides that did not meet additional stringent cut-off criteria (Andromeda score <40 or site localization probability<0.75). For each group of samples, the MS intensity results of two technical replicates for each biological replicate were combined for analyses. The MS intensity ratios 1 h/0 h(FC1), 6 h/0 h (FC6), and 12 h/0 h (FC12) were calculated,and compared between Rp1 and Rp2. For the determination of time-course regulation of each Ub site,the threshold values of 1.5 for up-regulation and 0.7 for down-regulation were adopted. Additionally, stringent criteria were established for the definition of up/down-regulation (Table S1). Ub sites or Ub proteins with consistent up/down-regulation (based on Rvalues) in Rp1 and Rp2 were defined as reproducibly quantified and only these sites or proteins were included for further quantification analyses,while the quantified Ub sites not fitting the criteria for reproducibly quantified substrates were not considered in time-course fold change analyses. The timecourse regulation of the corresponding qUb proteins containing reproducibly quantified qUb sites was determined according to the criteria in Table S1. The same criteria were also applied to the 6 h/1 h, 12 h/1 h, and 12 h/6 h ratios. A summary of number of Ub sites or Ub proteins included in each step of data processing has been provided in Figure S7.
Bioinformatics and statistical analyses
GO enrichment analyses were performed for each of the three GO categories: BP, MF, and CC. We used the subset of plant slim terms (http://geneontology.org) with the B73 maize genome as the background. GO annotations were downloaded from Ensembl (Ensembl Plants release 25, http://plants.ensembl.org). MapMan functional category enrichment analyses were carried out using the Zea mays L. genome release 1.1(http://mapman.gabipd.org/web/guest/mapmanstore) with the B73 genome as the background. The enrichment analyses in this study were carried out using the topGO R language package (Adrian Alexa, Jorg Rahnenfuhrer, http://www.bioconductor.org). The q value was calculated as described previously [68].
The online tools motif-x (http://motif-x.med.harvard.edu/motif-x.html) [34,35] and iceLogo (http://iomics.ugent.be/icelogoserver/create) [36] were used for identifying consensus ubiquitylation motifs. In motif-x, the ubiquitylation motif window was set to 13 residues, occurrences were set at 20 and significance was set at 1×10-6.The sequences of 1880 unique ubiquityl-13-mers that were located upstream of the 6th residue from the protein’s C terminus were used as the foreground for motif-x analysis. The B73 maize proteome (UniProt:UP000007305)was used as the background.The heatmap representation of the -6 to +6 residues surrounding the ubiquitylated lysine was generated with iceLogo using the 1926 unique Ub sites with protein sequence information. The precompiled Swiss-Prot composition of Z. mays L. was used as the reference and the P value was set as 0.05.
Interactions between all identified Ub proteins or qUb proteins were identified using the online tool STRING 10 (http://string-db.org/) [36], with the minimum required interaction score set as ≥0.7. All other parameters were set as default,and all active prediction methods were used. The networks were visualized using Cytoscape 3.2.1 software(http://www.cytoscape.org/),using the default preferred layout‘‘prefuse force directed layout”. Manual adjustments were made to reveal overlapping nodes and to fit the network to a compact scale for display.The interaction network of the regulated qUb proteins was generated using the same method and settings.There was discordance between the external node IDs output by STRING 10 and the original input protein IDs at the protein isoform level, resulting in minor alterations in the functional category annotations of the node proteins(~2% ).In this article,all analyses are based on the annotations corresponding to the STRING 10 output external node IDs, for details about the discrepancies in protein IDs, please see Data S3 and Figure 5A-C. In the network of regulated qUb proteins, the type of regulation (i.e., whether the qUb is classified as Up, Down,or Others) corresponds to the original input protein IDs (see Data S8 for the text summary and Data S9 for the outputinput discrepancies). The analyses of node degrees were performed using R language packages (https://www.R-project.org). Hierarchical clustering analyses were performed using the R language package ‘‘heatmaply”.
Data availability
All raw MS data and MaxQuant output results for the Ub sites and Ub proteins in this study are available at http://proteomecentral.proteomexchange.org/cgi/GetDataset as ProteomeX change:PXD007880.The MS proteomics data utilized to confirm the Ub sites and Ub proteins with altered levels of ubiquitylation (Data S5 and S6) are available as ProteomeXchange: PXD012897.
Authors’ contributions
BCW, XHL, YFW, and QC conceived this study and participated in its design. YFW performed statistical analyses. QC provided technical help. ZL constructed the proteome database translated from the RNA-seq data and conducted statistical analyses. TCL processed proteomics data and wrote the sample preparation and MS data analyses methods in the Materials and methods section of the manuscript. HYZ and CFZ performed the MS experiments.ZS prepared plant material and performed protein extraction. YFW wrote the manuscript together with TCL. QC revised the manuscript with input from ZL and ZS.All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by the National Key R&D Program of China(Grant No. 2016YFD0101003), the ‘‘Strategic Priority Research Program” of the Chinese Academy of Sciences(Grant No. XDA08010206), and the Agricultural Science and Technology Innovation Program of Jilin Province ‘‘Discovery of excellent germplasms and cultivation of inbred lines suitable for mechanized harvesting in maize” (Grant No.CXGC2017JQ019).We thank Dr.Zhifang Gao for generating the images of the hierarchical clustering analysis results.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2018.05.005.
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Intriguing Origins of Protein Lysine Methylation:Influencing Cell Function Through Dynamic Methylation
- Tung Tree (Vernicia fordii) Genome Provides A Resource for Understanding Genome Evolution and Improved Oil Production
- Multi-omics Analysis of Primary Cell Culture Models Reveals Genetic and Epigenetic Basis of Intratumoral Phenotypic Diversity
- Meta-analysis Reveals Potential Influence of Oxidative Stress on the Airway Microbiomes of Cystic Fibrosis Patients
- Schizophrenia-associated MicroRNA-Gene Interactions in the Dorsolateral Prefrontal Cortex
- A Computational Approach for Modeling the Allele Frequency Spectrum of Populations with Arbitrarily Varying Size