Sigma因子6促进拟南芥MIRNA基因转录
2019-05-10杨天宇郑丙莲
杨天宇,郑丙莲
(1.复旦大学 遗传工程国家重点实验室,上海 200438; 2.复旦大学 生命科学学院,上海 200438)
MicroRNA(miRNA)是一类长度在20~24nt的单链内源小分子RNA[1],广泛存在于真核生物中,由Victor Ambros等于1993年在线虫(Caenorhabditiselegans)[2]中首次发现.植物中的miRNA被发现的时间较晚,2002年,陈雪梅[3]实验室和David Bartel[4]实验室才在拟南芥(Arabidopsisthaliana)中发现miRNA的存在.miRNA能够通过切割靶标的mRNA或者抑制翻译达到在转录后水平调控基因表达的目的.在拟南芥中,miRNA的生物合成途径已经被了解得非常清楚.首先,DNA依赖的RNA聚合酶Ⅱ(PolⅡ)[5]将MIRNA基因转录成具有茎-环结构的初级miRNA(pri-miRNA)[6];pri-miRNA被Dicer like 1(DCL1)[7]蛋白主导的复合体进行第一次加工切割,成为前体miRNA(pre-miRNA);pre-miRNA被DCL1蛋白复合体再次切割形成成熟的miRNA/miRNA*双链[8].双链miRNA被HUA ENHANCER 1(HEN1)甲基化后[9],其中的一条链与ARGONAUTE1(AGO1)蛋白结合[10],参与到后续的效应途径[11].
尽管人们对miRNA生物合成途径的认识已经比较完善,但对部分环节的理解仍然不是十分清楚.比如PolⅡ转录MIRNA基因是否受到特定转录因子的调控?我们通过EMS(甲基磺酸乙酯)诱变DCL1蛋白功能部分降低的拟南芥弱突变体dcl1-14,在筛选miRNA水平异常的突变体的过程中,我们分离得到了一个新的突变体m112.本研究克隆了M112基因,并对M112在miRNA产生途径中发挥的功能展开研究,完善对植物miRNA生物合成途径的了解.
1 材料与方法
1.1 材料
模式生物为拟南芥(Arabidopsisthaliana),其中野生型植株包括Col-0(Columbia)和Ler(Landsberg erecta)两种生态型,突变体植株包括m112、sig6-2、sig6-3、sig2-2、sig3-2、sig4、sig5等(均为Col-0生态型背景),转基因植株包括MIRNA基因(MIR390a、MIR778、MIR172a1)启动子驱动的GUS转基因植株,以上植株材料均由本实验室前期构建或由其他课题组惠赠.大肠杆菌E.coliDH5α、农杆菌AgrobacteriumtumefaciensGV3101、大肠杆菌过表达载体pEarleyGate101(经过改造,去掉了GFP的标签)、大肠杆菌过表达载体p35S∶∶C3F(改造自pEarleyGate202,将原来的1×FLAG换成了3×FLAG),由本实验室前期保存.所用引物或探针由上海生工生物工程有限公司合成.其他实验所用试剂为国产或进口分析纯.
1.2 方法
1.2.1 图位克隆
将突变体m112(Col-0生态型背景)与野生型(Ler生态型)进行杂交,得到F1子代,F1代自交得到F2代,挑取F2代中表型为m112表型的植株作为样品进行编号,另外准备Col-0、Ler及Col-0/Ler杂合植株作为参照组,提取基因组DNA.分别以上述基因组DNA为模板,使用特定的标签引物[12]进行PCR反应.分析每个样品的电泳结果,如果出现与参照组Col-0、Ler、Col-0/Ler一致的结果(条带大小一致),分别记为0、2、1点.统计每个标签上所有样品的点数总和,除以样品总数,再除以2,即可得到每个标签对应交换率,从而将突变基因M112锁定在交换率为0的标签区域上.
1.2.2 全基因组测序分析及Sanger测序验证
将突变体m112的基因组DNA送到晶能生物技术公司进行全基因组测序,然后对图位克隆后锁定的区域进行分析,同时结合m112的表型以及TAIR网站上对于各个可能基因的描述,推测突变基因.为了验证猜测结果,我们提取m112的基因组DNA,并使用相应的引物(表1)对推测基因所在的区段进行Sanger测序,再将测序的结果与Col-0相对应的序列进行碱基比对,判断在m112中该基因是否发生了无义突变或错义突变.
1.2.3 等位分析与转基因互补
在初步确认m112的突变基因之后,为了进一步验证其准确性,我们拿到了M112等位基因已知的突变体,并将其与m112进行杂交,收种后得到F1代的种子,将F1代的种子播下以后,观察其表型,若表型与m112一致,则说明判断正确,反之则判断错误.另外,我们将M112构建到p35S∶∶(无tag)和p35S∶∶C3F载体上,通过GV3101侵染M112基因的突变体,拿到纯合且单拷贝插入的植株后,观察其表型,若表型与Col-0一致,则说明判断正确,反之则判断错误.
1.2.4 Northern blot

使用TRIzol溶液提取目标实验材料中的总RNA,然后使用5mol/L NaCl溶液和50% PEG8000溶液富集小分子RNA.接着将小分子RNA进行聚丙烯酰胺尿素凝胶电泳,并使用转膜仪将小分子RNA转到尼龙膜上,紫外交联并烘干,随后使用小分子RNA探针(表2)杂交过夜.第2天杂交结束后进行洗膜、封闭、染色、洗膜等一系列操作,最后显色并记录实验结果.
1.2.5 小分子RNA深度测序
我们收集Col-0和m112的花序材料,送到晶能生物技术公司对其进行小分子RNA的深度测序,共计两批样品即两次生物学重复.标准化后的数值使用RPTM(reads per ten million)进行计算,且筛选reads数大于100的miRNA表达数据进行统计,分析结果.
1.2.6 荧光定量PCR
使用TRIzol溶液提取目标实验材料中的总RNA,然后依次用DNaseⅠ、水饱和酚和氯仿对其进行纯化.接着使用OligodT引物、dNTP、DTT和RNA反转录酶将纯化后的总RNA反转录为cDNA,再加入SYBR Green supermix和对应的pri-miRNA引物(表3)进行荧光定量PCR.反应结束后,对SYBR荧光表达数据进行分析.

表3 qPCR所用引物
1.2.7 GUS染色
将MIRNA基因启动子驱动的GUS转基因植株与M112基因的突变体进行杂交,得到纯合的转基因突变体植株.然后取其花序、幼苗或者叶片等材料,放入加有适量GUS染色液的EP管中.接着将EP管放入真空泵中,打开EP管的盖子,抽真空5min,使得拟南芥材料完全没入到GUS染色液中.再将EP管放到37℃恒温培养箱中进行染色.染色完成后倒掉染色液,用75%乙醇清洗3次,最后在体式显微镜下观察并记录染色结果.
2 结果与分析
2.1 突变体m112的突变基因为SIG6
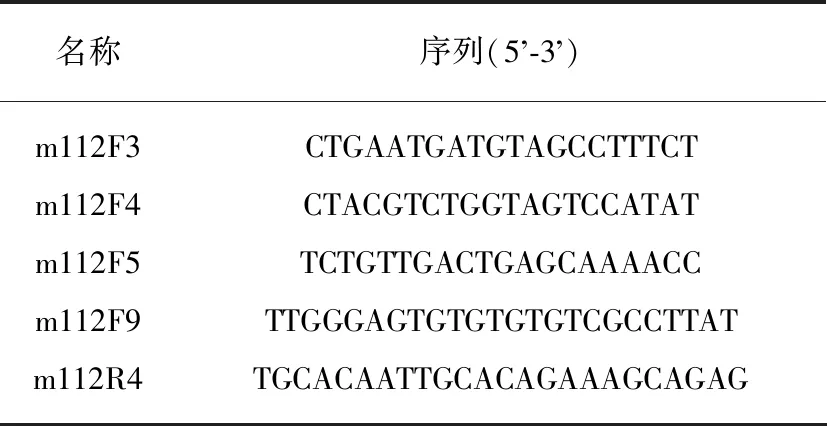
在筛选拟南芥miRNA水平异常的突变体的过程中,我们分离得到了一个新的突变体m112,它在发育早期,会出现子叶黄化的表型,且生长发育较野生型缓慢.通过图位克隆,我们将突变基因M112锁定在拟南芥第2条染色体15294K~15582K的区段中,如图1(a)(见第172页)所示.随后,我们将m112的基因组DNA进行全基因组测序,并对上述区段之间所有编码基因序列进行分析,结合m112的表型以及TAIR网站上对于各可能基因的描述,我们猜测突变基因为AT2G36990即SIG6.同时Sanger测序验证了在m112中,SIG6基因的第1680位碱基发生了C→T的突变,使得该基因的表达提前终止,即无义突变,如图1(b)所示.
目前,关于SIG6基因已报道的突变体sig6-1和sig6-2分别是由于在SIG6基因第4个、第5个外显子内插入了T-DNA而导致基因无法正常表达,而突变体m112则是由于碱基突变,使其SIG6基因在第6个外显子的末端表达终止,导致后3个外显子缺失,如图1(c)所示.为了后续研究的开展,我们得到了sig6-2及其他sigma因子突变体的种子.我们发现sig6-2和m112两者的形态学表型相似,在发育早期子叶会出现黄化,而将两者进行杂交,F1代仍然会出现黄化的表型,同时,我们将SIG6过表达质粒通过GV3101侵染到sig6-2中,发现纯合且单拷贝插入的植株不再出现黄化的表型,从两方面证明m112的突变基因确实是SIG6,如图1(d)、(e)所示.
图1 突变基因M112的鉴定及突变体表型观察Fig.1 Identification of the mutated gene M112 and observation of mutants(a) 突变基因M112在拟南芥第2条染色体上各引物标签所对应的交换率,在F1O11和T2N18 2个标签之间的区段交换率为0;(b) 突变体m112的Sanger测序峰图,图中淡蓝色框选中的碱基为突变碱基,该位点的碱基在野生型中为C,在突变体中突变为T;(c) sig6突变体的基因组示意图,其中sig6-1和sig6-2是已报道的sig6突变体,分别在第4个、第5个外显子中插入了T-DNA,m112则是在第6个外显子的末端表达终止,红色*表示m112的点突变位置;(d) Col-0、sig6-2、m112、sig6-2与m112的杂交F1代及SIG6转基因过表达植株的苗期表型;(e) Col-0、sig6-2及m112的发育后期表型,其中Ⅰ为整体植株对比,Ⅱ为花序对比,Ⅲ为叶片对比.
2.2 SIG6参与了miRNA的生物合成
为了确定sig6突变体中miRNA的累积是否出现了下调,我们首先进行了Northern blot实验.我们收集了包括Col-0、sig6-2、m112以及其他一系列sigma突变体的花序材料,在提取总RNA并富集小分子RNA后,进行了Northern blot实验,分别检测了U6(作为内参)及miR168、miR159、miR167、miR171、miR319、miR165/6和miR173的表达量,如图2(a)(见第173页)所示,发现在SIG6基因的两个不同性质的突变体中,所检测miRNA的累积量相比于Col-0均出现了不同程度的下调.
为了观察在突变体sig6中miRNA的整体水平是否产生了变化,我们收集了两批Col-0和m112的花序材料,进行两次生物学重复的小分子RNA深度测序.测序后将标准化的数值使用RPTM(reads per ten million)进行计算,且筛选reads数大于100的miRNA表达数据进行统计并分析结果.最终发现突变体m112的miRNA累积相比于野生型有明显地下调,如图2(b)所示,盒图中线的纵坐标数值接近-1即整体下调了近一半.此外,我们具体分析了第1批测序结果中miR168、miR159、miR167、miR171、miR319、miR165/6和miR173的表达量,发现m112中的这些miRNA相比于Col-0均有明显的下调(但对于miRNA链的选择并没有受到影响),如图2(c)所示,与之前的Northern blot结果形成了相互印证.以上实验结果证明突变体sig6中miRNA的累积下调,即SIG6在miRNA的生物合成中发挥作用.
2.3 SIG6影响了MIRNA基因的转录
为了探究SIG6具体影响了miRNA生物合成的哪一个步骤,我们首先检测sig6突变体中pri-miRNA的累积量是否发生了变化.我们提取了Col-0、m112、sig6-2、hyl1-2的总RNA(HYL1会参与到pri-miRNA的后续加工中,因此突变体hyl1-2中pri-miRNA会累积,这里作为正对照组),经过纯化和反转录得到各自的cDNA,并进行了两组荧光定量PCR(qPCR)实验,使用的引物为内参UBQ5及各个pri-miRNA对应的引物.结果发现相比于Col-0,m112和sig6-2中各个pri-miRNA的累积均出现了下调,如图3(a)、(b)(见第174页)所示.由此我们猜测在sig6突变体中,MIRNA基因的转录受到了抑制或者pri-miRNA的降解速度变慢了.
为了验证上面的猜测是否正确,我们将MIRNA基因MIR390a、MIR778、MIR172a1启动子驱动的GUS转基因植株杂交到sig6-2突变体的背景下,然后进行GUS染色,并与Col-0背景下的染色结果进行比较.如果是MIRNA基因的转录受到了影响,预期将看到sig6突变体中GUS的染色变浅.结果我们发现在相同的染色条件下,sig6-2突变体的背景下GUS染色强度均明显弱于Col-0背景下的GUS染色强度,如图3(c)所示.表明在sig6-2突变体中,MIRNA基因的转录受到了抑制,亦即MIRNA基因启动子的活性受到了SIG6的正调控.
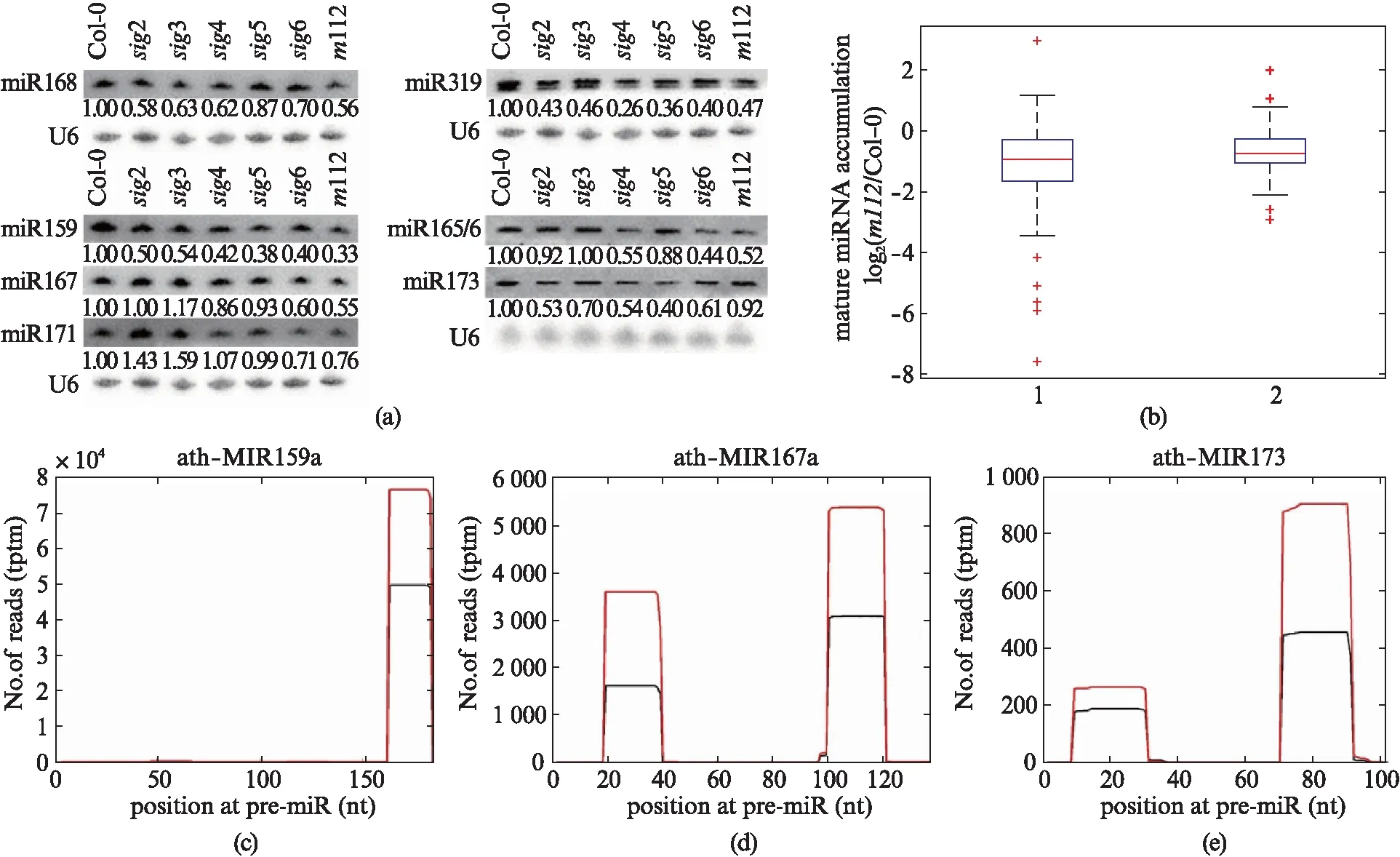
图2 Northern blot及小分子RNA深度测序检测sig6突变体中miRNA水平的变化Fig.2 Northern blot and small RNA sequencing to detect the changes of miRNA levels in sig6 mutants(a) Northern blot检测Col-0、sig6-2、m112以及其他sigma突变体中部分miRNA的累积量,每张膜从左至右的样品依次为Col-0、sig2、sig3、sig4、sig5、sig6和m112,使用的探针包括U6及miR168、miR159、miR167、miR171、miR319、miR165/6、miR173;(b) 小分子RNA深度测序分析Col-0和m112中成熟miRNA的水平,盒图中纵坐标是计算log2以后的m112/Col-0比值,横坐标的1、2分别指第1、2批测序样品(花序RNA);(c) 第1批测序样品中Col-0与m112的miR159a的表达量比较,其中红线为Col-0,黑线为m112;(d) 第1批测序样品中Col-0与m112的miR167a的表达量比较,其中红线为Col-0,黑线为m112;(e) 第1批测序样品中Col-0与m112的miR173的表达量比较,其中红线为Col-0,黑线为m112.

图3 qPCR检测sig6突变体中pri-miRNA的变化及不同背景下的proMIR∶∶GUS染色结果观察Fig.3 qPCR to detect the changes of pri-miRNA levels in sig6 mutants and observation of proMIR∶∶GUS staining in different background(a) 荧光定量PCR检测样品中的pri-miRNA水平,其中所用材料为Col-0、m112以及hyl1-2,hyl1-2作为正对照,UBQ5作为内参,检测引物为pri-miR171a、pri-miR172a、pri-miR167a、pri-miR156a、pri-miR159a、pri-miR166a、pri-miR168a、pri-miR319a共8组,误差线的数值为3次重复实验的STDEV计算值;(b) 荧光定量PCR检测样品中的pri-miRNA水平,其中材料为Col-0以及sig6-2,UBQ5作为内参,检测引物为pri-miR167a、pri-miR165a、pri-miR173a共3组,误差线的数值为3次重复实验的STDEV计算值;(c) proMIR∶∶GUS染色结果显示SIG6影响MIRNA基因启动子的活性,使用的转基因植株包括MIR390a∶∶GUS、MIR778∶∶GUS和MIR172a1∶∶GUS,实验材料均为6 d的光下苗,GUS染色时间为5h.
3 讨 论
拟南芥拥有超过300个MIRNA基因,从影响MIRNA基因整体转录的角度来看[13],TATA盒的核心启动子元素和至少21个顺式调控的motif被聚集到MIRNA的启动子序列上[14],暗示了整体上MIRNA的转录受到很多转录因子的调控.MAC(MOS4-ASSOCIATED COMPLEX)是目前研究较多的影响MIRNA基因转录的蛋白复合体,在拟南芥中具有抵御外界环境和调控生长发育的功能,其组分包括CDC5、PRL1、MAC7和MAC3等蛋白.DNA结合蛋白CDC5(a MYB-related protein)能够结合到MIRNA基因上促进PolⅡ的转录[15];转录因子PRL1(PLEIOTROPIC REGULATORY LOCUS 1)是一种保守的WD-40蛋白,能够与CDC5、DCL1、PolⅡ等蛋白产生互作,通过与pri-miRNA发生相互作用来使得miRNA的加工复合体更稳定[16];MAC7[17]、MAC3[18]和PRL1类似,不影响MIRNA启动子的活性和pri-miRNA转录本的半衰期,但能够通过影响HYL1的定位或促进DCL1的活性来使得pri-miRNA更稳定.目前,人们认为MAC作为一个复合体,可以通过调控pri-miRNA的形成、加工和稳定性来控制成熟miRNA的水平.除了MAC以外,常规的转录辅激活剂Mediator能够使得PolⅡ被招募到MIRNA基因的启动子中,从而触发真核生物中PolⅡ的转录激活[19].NOT2(NEGATIVE ON TATA LESS 2)能够与PolⅡ的C端结构域发生相互作用,从而促进PolⅡ的转录功能[20].细胞周期蛋白依赖的蛋白激酶(包括CDKF1和CDKDs等)也能够通过介导调节PolⅡ大亚基的C末端结构域(RNAPII-CTD)的磷酸化水平,起到调控MIRNA基因转录的作用[21].而我们的研究则发现在拟南芥中,质体Sigma因子6能够通过影响MIRNA基因启动子的活性去调控miRNA的生物合成.
目前已有的关于SIG6的研究表明,SIG6是Sigma因子家族中的一员,存在于质体中,主要通过调控质体基因的表达来参与叶绿体的早期发育[22].但是在拟南芥中,MIRNA基因的转录乃至pri-miRNA的后续加工均发生在细胞核中,那么存在于质体中的SIG6是如何参与到miRNA的生物合成途径中的呢?又如何能够调控MIRNA基因启动子的活性?我们据此提出了两种假设: 一是SIG6在质体中能够产生逆行信号反向地去调控细胞核的基因表达,即miRNA的产生可能受到质体与细胞核的相互交流的调控,提示原核细菌衍生的叶绿体的基因转录体系可能在早期miRNA的进化上具有一定的作用;二是猜测SIG6不仅仅存在于质体中,在细胞核中也有一定的分布,并通过影响PolⅡ对MIRNA基因的转录调控miRNA的合成.这两种假设的正确性需要我们进一步地进行探究.总体来说,本研究的意义在于提供线索证明质体因子能够以某种机制影响细胞核基因的表达,从而为研究miRNA生物合成途径提供了新的思路和视角.
致谢:感谢上海师范大学黄继荣老师课题组提供的sigma突变体种子.
