Mixed serous-neuroendocrine neoplasm of the pancreas:A case report and review of the literature
2019-04-22YueMeiXuZhiWenLiHongYanWuXiangShanFanQiSun
Yue-Mei Xu, Zhi-Wen Li, Hong-Yan Wu, Xiang-Shan Fan, Qi Sun
Yue-Mei Xu, Zhi-Wen Li, Hong-Yan Wu, Xiang-Shan Fan, Qi Sun, Department of Pathology,Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School,Nanjing 210008, Jiangsu Province, China
Abstract
Key words: Mixed serous-neuroendocrine neoplasm; Pancreatic serous cystic neoplasm;
INTRODUCTION
Pancreatic serous cystic neoplasms (PSCNs) account for only 1%-2% of all pancreatic tumors[1].In the 2010 World Health Organization (WHO) Digestive System Tumor Classification, PSCN was classified into five subtypes[2], including serous microcystic cystadenoma (SMA), serous oligocystic cystadenoma (SOA), solid serous adenoma,Von Hippel-Lindau (VHL) syndrome-associated serous cystic neoplasm, and mixed serous-neuroendocrine neoplasm (MSNN).MSNNs are termed mixed tumors containing two components with different pathologies, namely, PSCN and pancreatic neuroendocrine tumor (PanNET).For MSNNs, PSCN diffuse involvement of the whole pancreas is extremely rare, with only eight cases reported in the literature[3-10].Here, we report a novel case of diffuse PSCN admixture with an isolated PanNET in a Chinese woman and review the previous literature.The timeline is shown in Table 1.
CASE PRESENTATION
Chief complaints
A 45-year-old Chinese woman, with no obvious symptoms, was found to have a pancreatic neoplasm in the physical examination and admitted to our hospital for further diagnosis.
History of past illness
The patient had a free previous medical history.
Personal and family history
She denied any special personal or family history of VHL syndrome or other disease.
Physical examination
Abdominal palpation revealed a painless, mobile mass in the epigastrium, and no abnormalities were observed in an examination of the nervous system and ocular system.
Laboratory examinations
Laboratory tests for blood glucose, amylase, and tumor markers, including carcinoembryonic antigen (0.51 ng/mL, normal 0-10 ng/mL), cancer antigen-19-9(11.11 U/mL, normal 0-27 U/mL), and cancer antigen-242 (8.54 U/mL, normal 0-10 U/mL) were within normal limits.
Imaging examinations

Table 1 Time line
A computed tomography scan showed multiple cystic lesions involving the whole pancreas ranging in diameter from 0.4 to 2 cm and also revealed an enhanced mass,2.2 cm in diameter, in the head of the pancreas (Figure 1A).Moreover, multiple cysts were found in the kidneys bilaterally (Figure 1B), and the right lobe of the liver contained a small cyst (Figure 1C).No abnormalities were noted on brain magnetic resonance imaging.
Further diagnostic work-up
Grossly, the pancreatic parenchyma was replaced with numerous discrete and confluent cysts.The pancreatic parenchyma displayed spongy-like changes on histological section, which was composed of cystic, honeycomb-like, and cysticnodular areas.The cysts varied in size, ranging from 0.1 to 1.1 cm in diameter.The nodular area was demarcated by grayish-white fibrous septa, and a radial scar was observed in the center of some nodules.Clear fluid was found in the cystic cavity.A grayish-yellow solid mass, which was 3.5 cm × 2 cm × 2 cm in size, was in the pancreatic head with a clear boundary separated from the cystic area (Figure 2).
Microscopically, the histological morphology of the diffuse cystic lesions and the solid mass appeared significantly different, and the two components of the pancreatic tumor were separated by compressed fibrous tissue without any overlapping zone(Figure 3A).The diffuse cystic area was composed of prominent microcystic serous cystadenomas lined with a single layer of cubic to flattened epithelium with clear cytoplasm; the nuclei were uniformly small, round-to-oval, and bland with dense homogenous chromatin (Figure 3B).The pancreatic duct was normal, without any communication with the cysts.The solid tumor in the head of the pancreas demonstrated a well-differentiated grade 2 PanNET:The tumor cells were arranged in a ribbon-like and trabecular architecture with fine chromatin, and the mitotic count was 1 per 10 high power fields (HPF) (Figure 3C).
Immunohistochemically, the lining epithelium of the diffuse serous cystadenoma stained positive for cytokeratin (CK) 7 (Figure 3D) and CK19.The low-grade PanNET was positive for synaptophysin, chromogranin A (Figure 3E), and CD56.Both of the two pancreatic tumors were negative for HER-2 (Supplementary Figure 1).The Ki67 labeling index of the PanNET was 5% (Figure 3F).In addition, the tumor cells were hormone-negative (including insulin, glucagon, and gastrin), which, along with the patient’s clinical presentation, suggested a nonfunctional PanNET.
FINAL DIAGNOSIS
Based on these analyses, a diagnosis of pancreatic MSNN was established, consisting of diffuse SMA with a concomitant grade 2 PanNET.With this diagnosis in mind, we suspected that the patient may have VHL syndrome as she had clinical manifestations associated with VHL syndrome[11]:Pancreatic MSNN, bilateral renal cysts, and a hepatic cyst.However, no evidence of hemangioblastoma of the central nervous system has been found in this patient, and she refused to take a genetic test to confirm theVHLgene mutation.
TREATMENT
A Whipple operation with total pancreatectomy and splenectomy was performed.
OUTCOME AND FOLLOW-UP
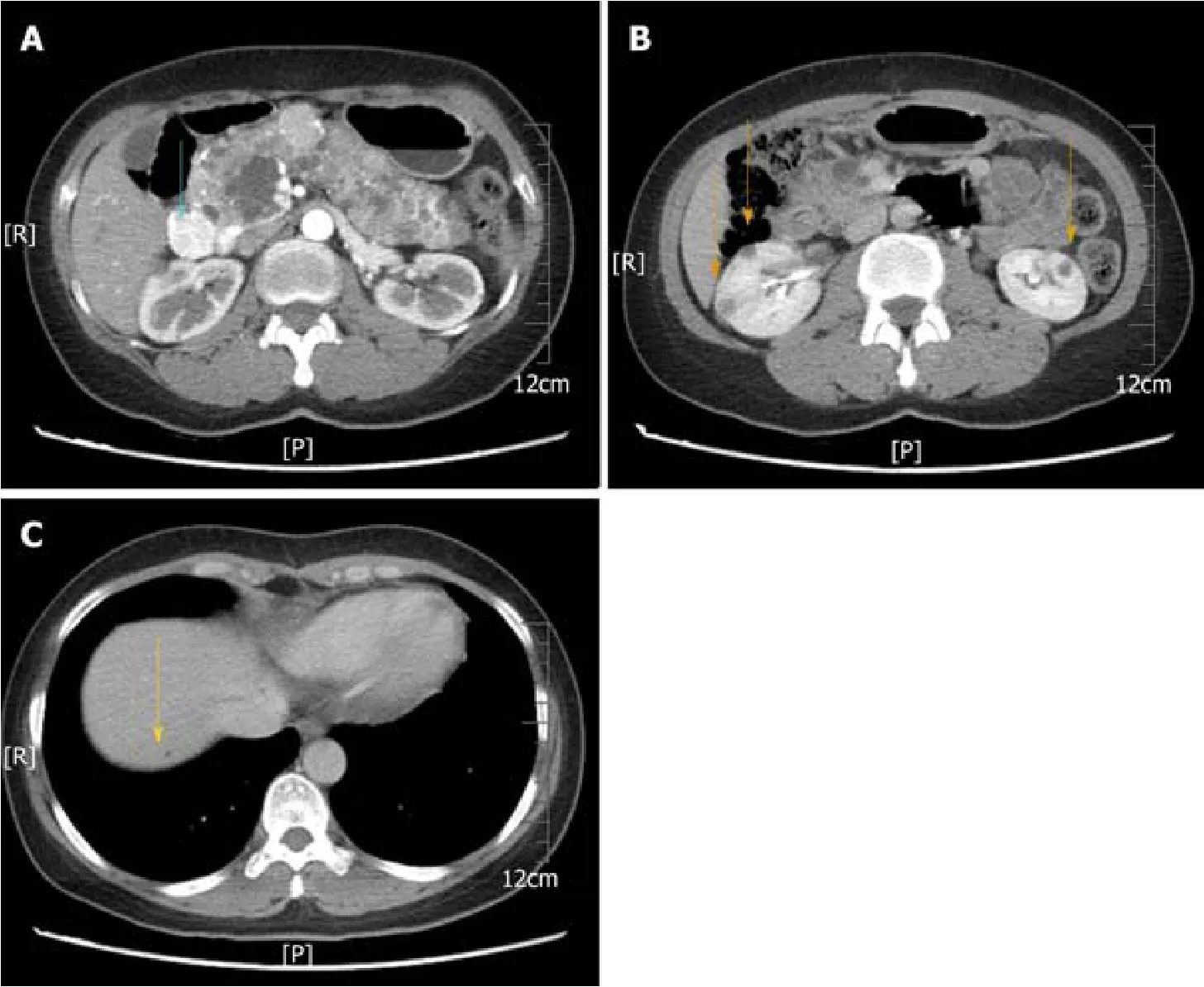
Figure 1 Abdominal computed tomography images.
After surgery, the patient was followed for 30 mo with no evidence of disease recurrence or metastasis of pancreatic tumor.
DISCUSSION
PSCNs are rare benign tumors.Occasionally, other tumors could be found concurrently with PSCN in an excised pancreatic specimen, such as PanNET,pancreatic ductal adenocarcinoma, intraductal papillary mucinous neoplasm, and metastatic tumors[12].PanNETs are the most common mixed tumor findings, and when found with PSCNs, the mixed tumor is termed MSNN according to the WHO classification[2].In a large cohort of 193 PSCNs, the simultaneous occurrence of PSCN and PanNET appeared in 6% (12/193) of all cases[12].Including the first report by Kameiet al[13]in 1991, 32 similar cases of MSNN were published in the Englishlanguage literature from PubMed (Table 2) until June 2019; however, only 22 cases,including ours, contain detailed clinicopathological data[3-10,13-22]to allow analysis.In the remaining specimens, the patients range in age from 25 to 78 years (mean, 50.1 years).There is a preponderance of females, with a female to male ratio of 19:3.The common tumor-related symptoms and signs include abdominal pain (7/22), nausea and vomiting (4/22), and obstructive jaundice (3/22), and one patient presented with watery diarrhea-hypokalemia-achlorhydria syndrome (WDHA) caused by a vasoactive intestinal polypeptide-secreting tumor (VIPoma)[21].Other patients were asymptomatic (8/22) or had rare clinical manifestations, including abdominal distension, postprandial fullness, melena, anorexia, peptic ulcer, and weight loss.

Figure 2 Photographs of the gross pancreas.
Although only a solitary PanNET component occurred in each analyzed MSNN case (Table 2), nine patients showed diffuse PSCN involving the whole pancreas, two had multiple PSCN, and the remaining 11 displayed an isolated PSCN.Most PSCNs were SMA or SMA with SOA component (15/22).SOA also appeared in five cases.Based on the distribution pattern of PSCN and PanNET, MSNN was categorized by Liet al[22]into four subtypes as follows:(1) Diffuse subtype:The entire pancreas is occupied by numerous PSCNs associated with one or more PanNETs, which could be located in any region of the pancreas; (2) Mixed subtype:The two different tumors combined with each other in a mass cannot be distinctly divided; (3) Solitary subtype:The two different components coincide in the pancreas without any intermingling area; and (4) Collision subtype:The two different lesions are separated from each other in most parts, but with a partially intermixed or overlapping zone.Among the analyzed MSNNs, 41% (9/22) were diffuse type, 27% (6/22) were mixed type, 23%(5/22) were solitary type, and 9% (2/22) were collision type.Histologically, most PSCNs can be easily differentiated from PanNETs.However, in rare cases, PSCN consists of small uniform nests or tubules with minimal lumen formation, specifically the solid variant of PSCN or solid serous adenoma[12].These cases can be misdiagnosed as PanNET[23].Indeed, the differential diagnosis of PSCNs and PanNETs can be challenging, especially in small biopsies.However, PSCN can be distinguished from PanNET based on the lack of reactivity for neuroendocrine markers (such as chromogranin A)[24], but it displays positive epithelial markers (such as CK19 and CK7) and possibly other markers (such as α-inhibin and calponin)[25,26].
The diagnostic nomenclature and classification criteria of PanNETs were inconsistent until 2010.However, the recent 2017 WHO classification classified pancreatic neuroendocrine neoplasms into well-differentiated NETs and poorly differentiated neuroendocrine carcinomas (NECs), as supported by genetic evidence[27].PanNETs are further graded on a scale of 1-3 based on mitotic rates and Ki67 proliferation index as follows:PanNET G1 with <2 mitoses/10 HPF and a <3%Ki67 index; PanNET G2 with 2-20 mitoses/10 HPF and a 3%-20% Ki67 index; and PanNET G3 with >20 mitoses/10 HPF and a >20% Ki67 index.Accordingly, after reviewing the pathological images of the previous case reports of MSNNs, all the PanNET components diagnosed as “low-grade/well-differentiated malignant NET”or “well-differentiated NEC” should be reclassified as well-differentiated PanNET rather than a poorly differentiated PanNEC.In the analyzed cases, PanNETs favored the pancreatic head/uncinate process region over the body and the tail (11:5:5) of the pancreas and ranged in size from 0.3 to 5 cm (mean, 2.2 cm).Although nine PanNETs were hormone-positive by immunohistochemical examination, including insulin,glucagon, vasoactive intestinal polypeptide, and somatostatin, all but one case presented with a nonfunctional PanNET.As mentioned before, a 69-year-old woman with a history of parathyroid adenoma presented with typical symptoms of WDHA syndrome, and then histological examination revealed a PSCN coexisting with a VIPoma in the tail of the pancreas.Although MEN-1 syndrome was suspected,molecular genetic analysis did not show aMEN-1gene mutation[21].
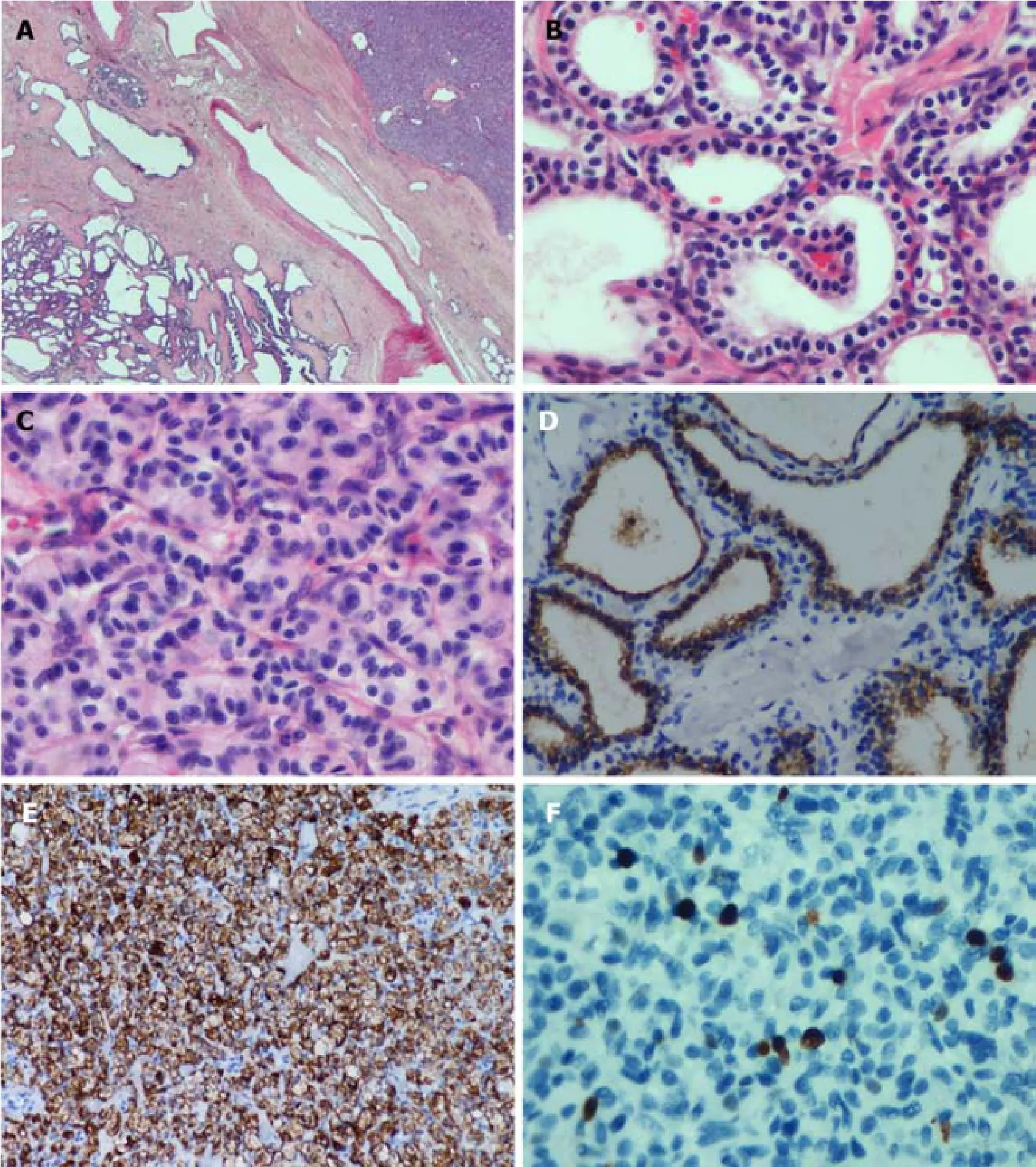
Figure 3 Histological morphology of the diffuse cystic lesions and the solid mass appears significantly different.
VHL syndrome is a rare autosomal dominant hereditary disease that is caused by mutations of the embryonic line of theVHLgene on chromosome 3p25-26[28].The incidence of pancreatic involvement in VHL disease is 17%-77%[8,29].PSCN and PanNET are classic presentations of VHL syndrome.In Hammelet al[29]’s study, 12.3%of VHL patients showed PSCN, 12.3% had PanNET, and 6.9% had MSNN.In addition, Cassolet al[30]reported that up to 17% of patients with VHL disease may develop PanNETs.In our reviewed cases from the literature, 14% (5/34) of MSNNs were confirmed to be associated with VHL syndrome, and two cases were suspected(Table 2).We categorized these patients into two groups according to the presence of VHL disease (Table 3).The mean age of VHL-related MSNN is less than that of the same VHL-unrelated association (33 yearsvs55 years,P< 0.001).MSNN with a diffuse PSCN component is more likely to occur in VHL patients (P= 0.043).It has been reported that nearly 10% of PSCN patients could be demonstrated to have a germline mutation of theVHLgene[31].Unfortunately, including our present case,none of the MSNNs reported previous genetic testing forVHLbeing performed, sothe incidence of MSNN in VHL patients is still unclear.Even though the tumor size of the PanNET component in MSNN is larger in VHL patients than in its non-VHL counterpart (P= 0.016), PanNETs demonstrated signs of violation behavior in both VHL and non-VHL groups, such as duodenal wall/peripancreatic soft tissue invasion, lymphatic/venous permeation, perineural invasion, and lymph node metastasis.Patients with VHL syndrome are prone to suffering visceral cysts and markedly vascularized neoplasms in various organs[28].As such, in addition to pancreatic tumors, patients in the VHL-related group were more likely to suffer from other tumors (P= 0.009), such as renal clear cell carcinoma, cerebellar/retina hemangioblastoma, liver angioma, adrenal pheochromocytoma, and paravertebral paraganglioma (Table 2).
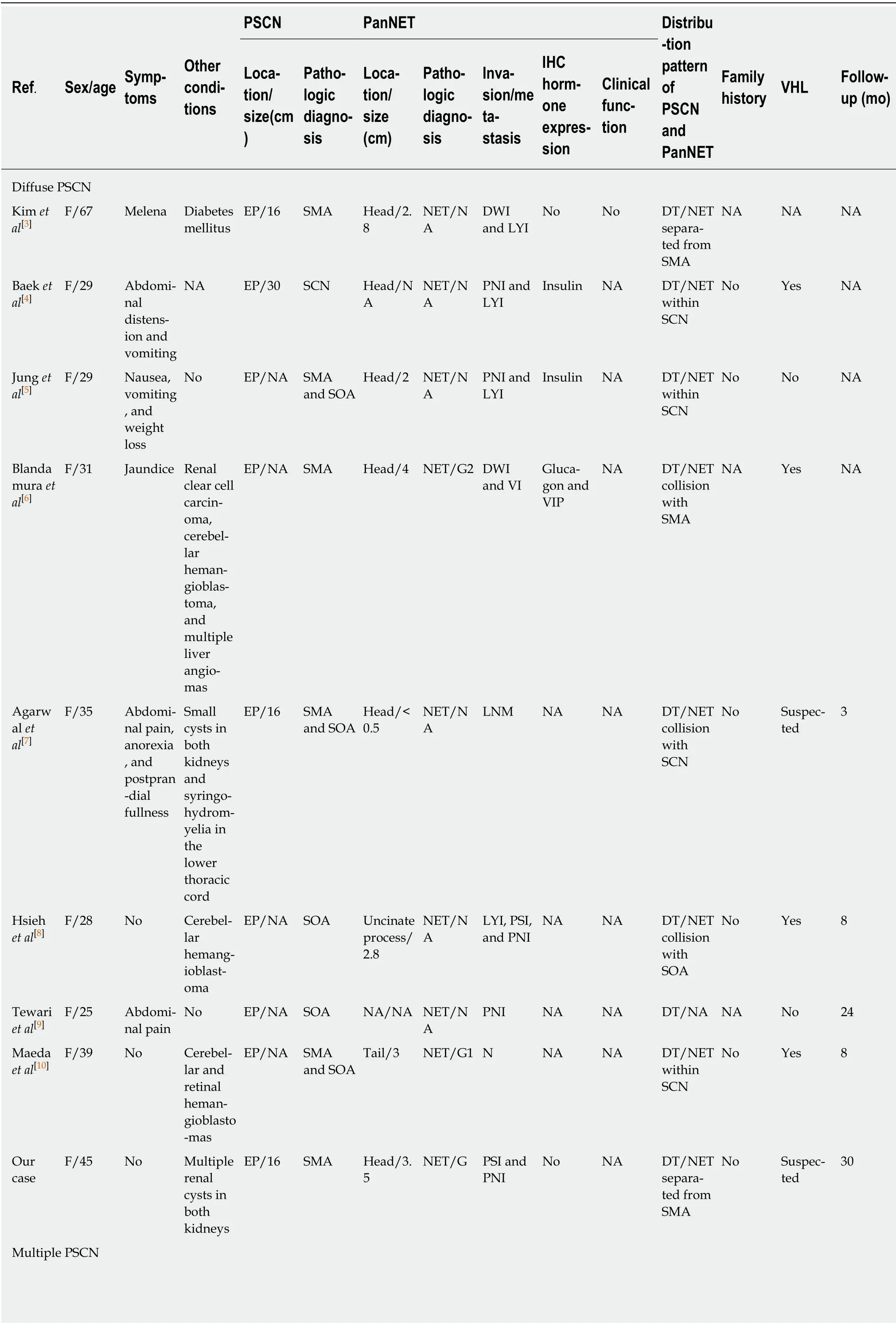
Table 2 Clinicopathological characteristics of mixed serous-neuroendocrine neoplasms

?

CT:Collision type; DT:Diffuse type; DWI:Duodenal wall invasion; EP:Entire pancreas; F:Female; LNM:Lymph node metastasis; LYI:Lymphatic invasion; MSNN:Mixed serous-neuroendocrine neoplasm; M:Male; MT:Mixed type; NA:Not available; NET:Neuroendocrine tumor; PNI:Perineural invasion; PSI:Pancreatic surrounding soft tissue invasion; ST:Solitary type; SMA:Serous microcystic cystadenoma; SOA:Serous oligocystic cystadenoma;SCN:Serous cystic neoplasm; VIP:Vasoactive intestinal polypeptide; VI:Venous invasion; WDHA:Watery diarrhea-hypokalemia-achlorhydria syndrome.
As all the PSCN components of MSNN are benign per our review, no recurrence of metastasis occurred during the follow-up period.Indeed, true malignant behavior is exceedingly rare in PSCN[12].Nevertheless, PanNETs are considered low-grade malignancies[2], so the prognosis of MSNN is dependent on the PanNET component.As demonstrated by Agarwalet al[7], lymph node metastasis occurred even when the diameter of the PanNET component was less than 0.5 cm.Therefore, the pathologist must perform a careful gross examination and sampling to exclude the presence of a concomitant PanNET in PSCN specimens.
There are two hypotheses to explain the formation of MSNN:(1) Common progenitor cells; or (2) Distinct progenitor cells.For the first hypothesis, both PSCN and PanNET could derive from a common pluripotent stem cell[15], as supported by the observation of biphasic differentiation in a pancreatic lesion, such as nesidioblastosis, a nonneoplastic disorder with coincident epithelial and endocrine differentiation[32].Additionally, Kakkaret al[14]identified neurosecretory granules,glycogen, and intermediate filaments within the same tumor cells by ultrastructural examination.This may explain cases in which both tumors are intermingled intimately but cannot explain those with separated lesions.For the second hypothesis,both counterparts in MSNN could derive from two different cell types, which would probably arise from a predisposing genetic disorder, such as VHL syndrome[6].Further support for this hypothesis is that the genetic mutations of PSCN are different from those of PanNET[31], indicating a distinctive pathogenesis of the two tumors.Taken together, further studies, especially molecular analysis, are still needed to elucidate the pathogenesis and origin of MSNN.
CONCLUSION
We report a rare case of MSNN with a diffuse PSCN component involving the entire pancreas in a Chinese woman.Due to its close relationship with VHL syndrome and the malignant potential of the neuroendocrine component, patients with MSNN should undergo thorough systemic examination, including molecular genetic analysis, and close clinical follow-up is recommended.
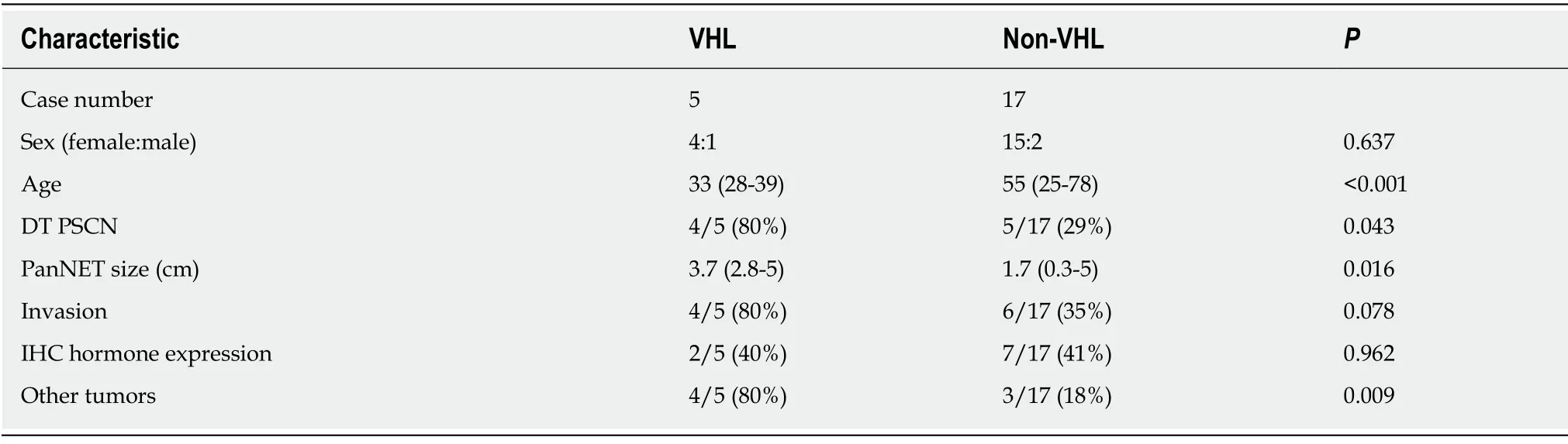
Table 3 Clinicopathological characteristics between Von Hippel-Lindau-associated and non-Von-Hippel-Lindau-associated mixed serous-neuroendocrine neoplasms
杂志排行

World Journal of Clinical Cases的其它文章
- Pure squamous cell carcinoma of the gallbladder locally invading the liver and abdominal cavity:A case report and review of the literature
- Management of massive fistula bleeding after endoscopic ultrasound-guided pancreatic pseudocyst drainage using hemostatic forceps:A case report
- Fatal complications in a patient with severe multi-space infections in the oral and maxillofacial head and neck regions:A case report
- Bouveret syndrome:A case report
- Left armpit subcutaneous metastasis of gastric cancer:A case report
- Rigid esophagoscopy combined with angle endoscopy for treatment of superior mediastinal foreign bodies penetrating into the esophagus caused by neck trauma:A case report