Aetiology of Legg-Calvé-Perthes disease: A systematic review
2019-03-21VitoPavoneEmanueleChisariAndreaVescioClaudioLizzioGiuseppeSessaGianlucaTesta
Vito Pavone, Emanuele Chisari, Andrea Vescio, Claudio Lizzio, Giuseppe Sessa, Gianluca Testa
Abstract BACKGROUND Legg-Calvé-Perthes disease (LCPD) is a clinical condition affecting the femoral head of children during their growth. Its prevalence is set to be between 0.4/100000 to 29.0/100000 children less than 15 years of age with a peak of incidence in children aged from 4 years to 8 years. LCPD aetiology has been widely studied, but it is still poorly understood.AIM To analyse the available literature to document the up-to-date evidence on LCPD aetiology.METHODS A systematic review of the literature was performed regarding LCPD aetiology,using the following inclusion criteria: studies of any level of evidence, reporting clinical or preclinical results and dealing with the aetiology or pathogenesis of LCPD. Two reviewers searched the PubMed and Science Direct databases from their date of inception to the 20th of May 2018 in accordance with the Preferred Reporting Items for Systemic Reviews and Meta-Analyses guidelines. To achieve the maximum sensitivity of the search strategy, we combined the terms: ‘‘Perthes disease OR LCPD OR children avascular femoral head necrosis” with “pathology OR aetiology OR biomechanics OR genetics” as either key words or MeSH terms.RESULTS We include 64 articles in this review. The available evidence on LCPD aetiology is still debated. Several hypotheses have been researched, but none of them was found decisive. While emerging evidence showed the role of environmental risk factors and evidence from twin studies did not support a major role for genetic factors, a congenital or acquired predisposition cannot be excluded in disease pathogenesis. One of the most supported theories involved mechanical induced ischemia that evolved into avascular necrosis of the femoral head in sensible patients.CONCLUSION The literature available on the aetiology of LCPD presents major limitations in terms of great heterogeneity and a lack of high-profile studies. Although a lot of studies focused on the genetic, biomechanical and radiological background of the disease, there is a lack of consensus on one or multiple major actors of the etiopathogenesis. More studies are needed to understand the complex and multifactorial genesis of the avascular necrosis characterizing the disease.
Key words: Legg-Calvé-Perthes disease; Aetiology; Pathogenesis; Genetics; Risk factors
INTRODUCTION
Legg-Calvé-Perthes disease (LCPD) is a complex disease affecting the epiphysis of the femoral head in the paediatric population. Historically considered an osteochondrosis, it is now being referred to as an idiopathic avascular necrosis of the femoral head in the paediatric population. Among its prevalence there is no general agreement. It is set to be between 0.4/100000 to 29.0/100000 children < 15 years of age with a peak of incidence in children aged from 4 years to 8 years and a male/female ratio of 5:1[1,2]. A high profile epidemiological study held in 2017[3]involving 2.1 million individuals attempted to report a more accurate prevalence of this disease. An overall prevalence of 9.3 per 100000 subjects was found. The male/female ratio was 3.1:1. Even though the study was conducted in Sweden from 1973 to 1993, it is one of the most up-to-date sources of evidence of LCPD epidemiology.
Despite the aetiology of the disease having been widely researched, it is still not fully understood. While the major hypothesis relies on a multifactorial genesis,several hypotheses involving mechanical, genetic and systemic conditions have been proposed to explain the pathogenesis of the femoral head osteonecrosis. The bestsupported theory involves interference with normal blood supply to epiphysis due to repetitive mechanical stress[4,5]. There is no consensus for the optimum treatment. The aim of treatment is to maintain the sphericity of the femoral head and the congruency of the femur-acetabulum relationship to prevent secondary degenerative arthritis,which eventually leads to total hip arthroplasty in 5% of cases[6]. Early diagnosis and management can help prevent the collapse of the femoral head, progressive femoral head deformity and impingement[7].
Children who have a skeletal age of 6.0 years or less at the onset of the disease do well without treatment[8]. Operative treatment should be considered in children who are 6 years old or older and have over 50% femoral head necrosis when the diagnosis is made[9].
MATERIALS AND METHODS
Literature search strategy
We conducted this systematic review according to the guidelines of the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA)[10]. A systematic review of two medical electronic databases (PubMed and Science Direct)was performed by two independent authors from their date of inception to May 20,2018. To achieve the maximum sensitivity of the search strategy, we combined the terms: ‘‘Perthes disease OR LCPD OR children avascular femoral head necrosis” with“pathology OR aetiology OR biomechanics OR genetics” as either key words or MeSH terms. The reference lists of all retrieved articles were reviewed for further identification of potentially relevant studies and assessed using the inclusion and exclusion criteria.
Selection criteria
Eligible studies for the present systematic review included those dealing with the aetiology of LCPD. The initial title and abstract screening was made using the following inclusion criteria: studies of any level of evidence, written in English,reporting clinical or preclinical results, published in peer review journals and dealing with the aetiology of LCPD. Exclusion criteria were articles written in other languages or studies with a focus on secondary/LCPD-like diseases caused by systemic conditions such as sickle-cell disease, inflammatory disease, the effects of chemotherapy, radiation or prolonged steroid use. We also excluded all the remaining duplicates, articles dealing with other topics, those with poor scientific methodology or without an accessible abstract. Reference lists were also hand-searched for further relevant studies. All publications were limited to in vivo, in vitro, animal and human studies in the English language. Abstracts, case reports, conference presentations,editorials and expert opinions were excluded.
Data extraction and criteria appraisal
All data were extracted from article texts, tables and figures. Two investigators independently reviewed each article. Discrepancies between the two reviewers were resolved by discussion and consensus. The final results and any remaining controversy on the reviewed article were reviewed and discussed with senior investigators.
Risk of bias assessment
In this systematic review, risk of bias assessment of the in vitro studies was not performed as there is no accepted grading scale for such studies. Risk of bias assessment of all in vivo selected full-text articles was performed according to the Regional Online Brownfields Information Network I for non-randomized studies[11].The Regional Online Brownfields Information Network I (ROBINS-I) tool consists of three stage assessment of the studies included. First stage regards the planning of the systematic review, the second stage is the assessment of the common bias possibly found in these studies and the latter is about the overall risk of bias (Table 1).
The assessments were performed by three authors independently. Any discrepancy was discussed with the senior investigator for the final decision. All the raters agreed on the final result of every stage of the assessment.
RESULTS
Included studies
A total of 1630 articles were found. After the exclusion of duplicates, 1078 articles were selected. At the end of the first screening, following the previously described selection criteria, we selected 130 articles eligible for full text reading. Ultimately, after full text reading and reference list check, we selected n = 64 articles following previous written criteria. A PRISMA[10]flowchart of the method of selection and screening is provided (Figure 1). The included articles[11-85]mainly focus on genetic research, epidemiological studies, magnetic resonance imaging analysis and histological histochemical analysis. The main findings of the included articles are summarized (Tables 1-4).
Smoking
While other environmental factors may be present, smoking seems to one of the most reported risk factor for developing LCPD[11-16]. In particular, Perry et al[12]recently showed how maternal smoking can affect the risk of developing the disease in a case control study. In addition to this, another four studies[13-16]report evidence of the association between environmental smoke and LCPD, both during maternalpregnancy and the childhood of the patient. Lastly, a study involving 852 patient showed how the smoking habit augmented the risk for LCPD by 100% in the examination sample[17].


LCPD: Legg-Calvé-Perthes disease; OR: Odds ratio; CI: Confidence interval; ADHD: Attention deficit hyperactivity disorder; IL-6: Interleukin 6; IGF:Insulin-like growth factor; IGFBP: Insulin-like growth factor binding protein; HR: Hazard ratio: HIF: Hypoxia-inducible factor.
Socioeconomical deprivation
Three different studies[14,18,19]involving an overall 340 LCPD patients explored the link between the disease and socioeconomic deprivation without revealing a significant association. On the contrary, Kealey et al[20], investigating 311 patients, found a higher prevalence among the most deprived rural category. In 2012, another study[21]analysed the incidence of LCPD between 1990 and 2008 in children between 0 years and 14 years in the United Kingdom. They reported how the incidence was coherently higher within the quintile with the highest degree of deprivation (risk ration = 1.49,95% confidence interval (CI): 1.10-2.04) (P < 0.01). Five more studies[22-26]held in United Kingdom by the same research group reported similar results.
Low birth weight
Metcalfe et al[27]reported an increased presence of low birth weight in children affected by LCPD. Similar to the results provided by Lappin et al[28], Sharma et al[18]reported an association between low birth weight and LCPD, whereas the weight of the children at the moment of the diagnosis and follow up did not show significant alteration. This was further supported by other studies with similar results[26]. On the contrary, Bahmanyar et al[17]reported a possible association between low birth weight and LCPD, but that was shown to be insignificant after evaluating other risk factors like maternal smoking. Only really low birth weight (< 1500 g) seemed to be associated independently with LCPD. Another nationwide study[29]held in Norway and involving 425 patients reported similar results. Their results supported the presence of environmental or genetic factors but not low birthweight. Similar results were reported by other epidemiological studies[12-16].
Attention deficit hyperactivity disorder (ADHD) and psychological burden
Loder et al[30]in 1993 studied the association between ADHD and LCPD in 24 patients.He reported the presence of one third (33%) of the children with abnormally high scores in profiles associated with ADHD. Perry et al[31,32]investigated both general practise registry of comorbidities in LCPD patients and the incidence of behavioural disturbance. ADHD was not associated with Perthes' disease (OR = 1.01, 95%CI: 0.48-2.12) in the first study[31]; while in the second one[32], a case control study involving 146 cases of LCPD and 142 hospital controls, the presence of behavioural disturbance was reported.
In 2015, a prospective study[33]evaluated in 58 adolescents patients (11 with LCPD)undergoing hip preservation surgery the presence of psychological disturbances. A significant presence of depression and anxiety symptoms was reported. Bergman et al[34]investigated ADHD in LCPD patients and in siblings of the child affected. A similar incidence was reported in both the groups. Hailer et al investigated the prevalence of the disease in two high profile studies held in 2012[35]and in 2014[36]. The first one involved 2579 LCPD patients and 13748 controls. It investigated the risk of injury in both groups and reported a higher percentage in the LCPD group. The second one involved 4057 individuals with LCPD in Sweden during the period 1964-2011 and 40570 individuals without LCPD randomly selected from the Swedish general population and matched by year of birth, sex and region. They reported that individuals with LCPD also had a raised hazard ratio (HR) of 1.5 (95%CI: 1.2-1.9) for ADHD.
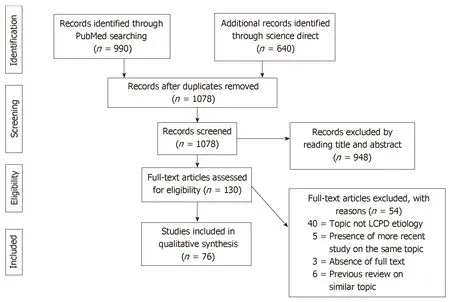
Figure 1 PRISMA flowchart of the systematic literature review. PRISMA: Preferred Reporting Items for Systematic Reviews and Meta-Analysis.
Türkmen et al[37], instead, did not find a significant difference in ADHD prevalence between LCPD group and control groups.
Obesity and leptin
A study held in 2016[38]reported a positive association between LCPD and obesity.Obesity was associated with a more severe clinical presentation and femoral head deformity. Lee et al[39]have investigated the levels of leptin in LCPD patients compared to control subjects. A significantly higher value concordant with the severity of the disease was reported.
Familiarity and genetic role
Over the years, several cases of LCPD in the same family have been reported[40,41]. This evidence suggested a genetic role in the development of the disease. In particular,Miyamoto et al[42]were the first to report a case of familiar LCPD associated with a mutation of the Collagen type II gene (COL2A1). Thus, COL2A1 genes were proposed as potential pathogenic trigger of LCPD. Several case reports also found evidence of this association in LCPD patients[43,44]. Further studies investigated the relationship between this gene mutation and LCPD. Su et al[45]recruited 42 members of a fivegeneration family and found in 16 patients a p.Gly1170Ser mutation of COL2A1 cosegregated with LCPD, precocious hip osteoarthritis or avascular femoral head necrosis not linked with LCPD. Li et al[46]in 2014 held a study, including a fourgeneration family, reporting the presence of the mutation in six affected family members.
Metcalfe et al[27]investigated the presence of genetic factors using the information derived from the Danish Twin Registry. After studying concordance with LCPD in 81 twin pairs (10 monozygotic, 51 dizygotic and 20 unclassified), they concluded that the absolute risk that a co-twin of an affected individual will develop LCPD is low, even in the case of monozygotic twin pairs. While Metcalfe et al[27]did not find an association between LCPD and birth weight, another twin study[28]and a case control study[17]involving an overall of 320 twin patients and 852 patients, respectively,concluded that low birth weight may play a role in developing the disease.
Coagulation disturbance
A meta-analysis[47]held in 2012 investigated factor V Leiden, prothrombin II and methylenetetrahydrofolate reductase (MTHFR) polymorphism as sources of possible genetic aetiology of LCPD. They comprised 12 case-control studies, including 824 children in the Perthes group and 2033 children in the control group. Factor V Leidenpolymorphism (carrying the minor A allele instead of the G allele) was associated with a three times higher incidence of the disease. Prothrombin II polymorphism (A allele instead of the G allele) reported giving a 1.5-fold increase risk of disease. There was no association between MTHFR polymorphism and Perthes disease. In 2015,Srzentic et al[48]investigated gene expression and variants by quantitative reversetranscriptase polymerase chain reaction and reported no difference between LCPD patients and controls for Factor V Leiden, Factor II, MTHFR and Plasminogen activator inhibitor-1.

Table 2 Main findings of included cohort studies


LCPD: Legg-Calvé-Perthes disease; ADHD: Attention deficit hyperactivity disorder; OR: Odds ratio: CI: Confidence interval; MRI: Magnetic resonance imaging.
Also, a prospective study held in 2002 did not suggest that thrombotic diatheses due to deficiency of protein C, protein S or antithrombin III or due to factor-V Leiden mutation are major causes of Legg-Perthes disease[49].
Inflammation markers
Liu et al[50]analysed age- and sex-matched serum samples from 10 control subjects and 10 patients with LCPD. They reported a higher presence of proteins and factors linked to complement and coagulation cascade. Increased activity of these factors may contribute to LCPD aetiology. In 2014 Srzentić et al[51]studied the association of frequencies of genetic variants of immune response genes with LCPD. They found significantly over-represented heterozygous subjects with an interleukin-6 (IL-6)polymorphism (G-174C/G-597A) in the LCPD group. In 2015, another studied reported a significantly increased IL-6 protein level in the synovial fluid of the hips affected by LCPD[52].
Apoptosis factors
Srzentić et al[48]investigated the expression of apoptosis genes by the quantitativereverse-transcriptase polymerase chain reaction technique in 37 patients. They reported a higher presence of proapoptotic factor Bcl-2-associated X protein (Bax)along with a significantly higher Bax/Bcl-2 ratio in the patient group. Zhang et al[49]found in a rat model similar results, with a higher expression of the apoptotic genes Casp3, Casp8 and Casp9 in chondrocytes after hypoxia.
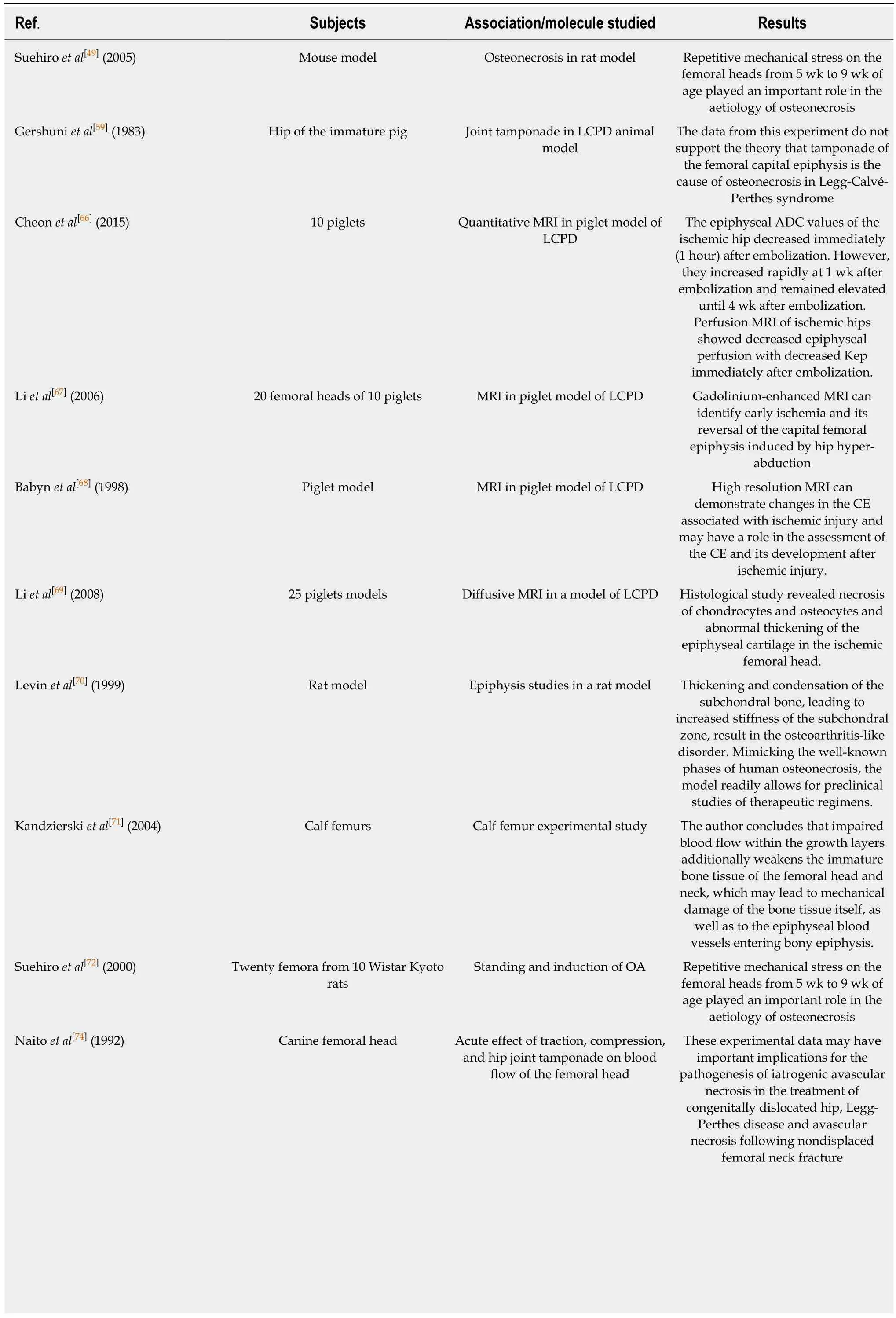
Table 3 Main findings of the included animal studies

LCPD: Legg-Calvé-Perthes disease; HIF-1: Hypoxia-inducible factor; VEGF: Vascular endothelial growth factor; MRI: Magnetic resonance imaging.
Mechanical stress and ischemia damage:Ischemia damage was reported in the first research documents we have on LCPD and its pathology insight[53,54]. In 1976, one of the first studies[55]about the aetiology of LCPD reported the presence of areas of infarction in 51% of hips histopathologically examined in 57 cases of femoral head biopsy. Calver et al[56]examined radiologically the ischemia areas in the hips of 50 children. The five with evidence of ischemia areas developed LCPD within 6 mo. The other 45 were healthy at 1 year follow up. Catteral et al[57]and Ponseti et al[58],respectively, in two different historical studies reported an area of ischemia in two children’s biopsies and morpho-structural alteration possibly associated with ischemic damage. In addition, several animal studies were conducted. A study held in 1983[59]investigated how joint tamponade in pigs would provoke ischemia of the epiphyseal plate. The necessity of high and prolonged pressure, which is difficult to achieve in normal conditions, was reported. Several research studies reported evidence of ischemia damage using radiological techniques[60-64]. This provides evidence of its role in the enteropathogenesis of the LCPD. Recently, Pinheiro et al[65]proposed a biomechanical model that further supports a combined role of both ischemic condition, skeletal immaturity and altered biomechanics. Also, studies on hip osteonecrosis in piglets confirmed this aetiology[66-69].
Mechanical stress-induced ischemia was found to be a possible aetiology of LCPD also using several animal models[70-72]and experimental analysis models[65,73,74]. One particular study performed by Suehiro et al[49]involving Wistar Kyoto rats reported how repetitive mechanical stress on the femoral heads from 5 wk to 9 wk of age played an important role in the aetiology of osteonecrosis. This theory is historically and currently one of the most supported[54,61,75-77].
Delayed epiphysial growth and hip geometry alterations:In 2003, Kitoh et al[78]investigated the epiphyseal height (EH) and width (EW) of the unaffected hip in 125 children affected by LCPD. A positive linear correlation (R = 0.87) was observed in the EH: EW ratio in these patients. A smaller EH than expected for EW in our series indicated epiphyseal flattening of the femoral head in LCPD cases. This finding supports the hypothesis that a delay in endochondral ossification in the proximal capital femoral epiphysis may be associated with the onset of Perthes' disease.
In 2014, Kocjančič et al[79]investigated the resultant hip force and contact hip stressdistribution in a population of 135 adult hips of patients who had been treated for LCPD. Contra-lateral hips with no record of disease were taken as control. The contact hip stress gradient index was 148% higher (P < 0.001); expressing an unfavourable,steep decrease of contact stress at the lateral acetabular rim. On the contrary, Pinheiro et al[80]reported how the anatomical variations appear to have only a limited effect on the stress distribution in the femoral epiphysis, even during high impact activities and in the presence of a skeletally immature epiphysis. However, the same group recently published a study[65]on a new biomechanical model and reported a possible involvement of vascular obstruction to the epiphysis that may arise when there is delayed ossification and when articular cartilage has reduced stiffness under compression.
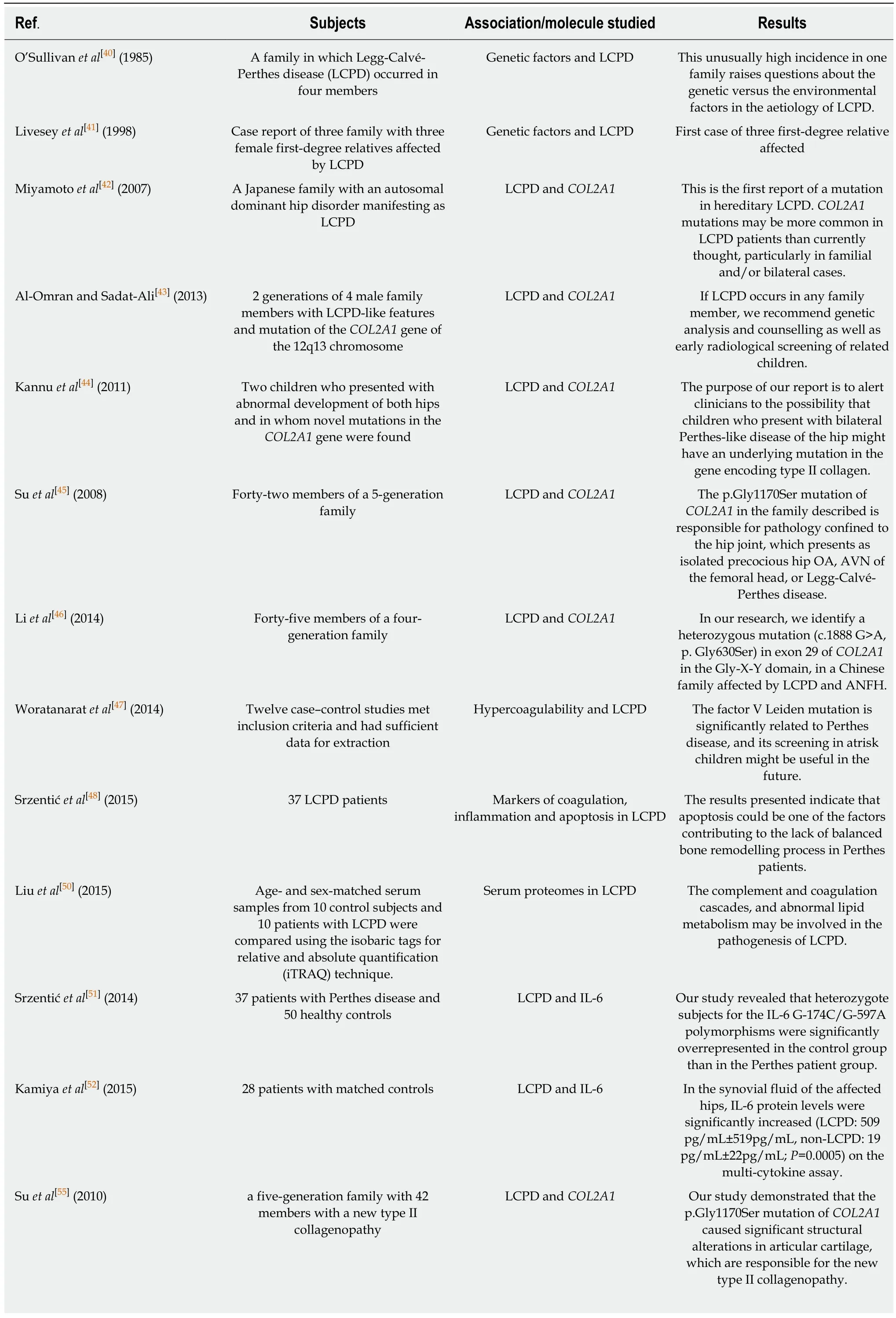
Table 4 The main findings of the included in vitro human studies are reported

LCPD: Legg-Calvé-Perthes disease; COL2A1: Collagen type II gene; IGF: Insulin-like growth factor.
Vascular endothelial growth factor (VEGF) and hypoxia-inducible factor (HIF-1):In 2004, an animal study[49]involving piglets with avascular necrosis reported increased VEGF protein and mRNA expression in the epiphyseal cartilage of the infarcted heads compared with the contralateral normal heads. Therefore, VEGF upregulation in the proliferative zone after ischemic damage was proposed as a possible stimulator of vascular invasion and granulation tissue formation. Zhang et al[81]reported also a higher expression of VEGF and HIF-1 in rat model chondrocytes.
VEGF was found to be low in LCPD patients in an Indian population-based study[49]. In 2014, 28 LCPD cases (mean age: 8 ± 3.8) and 25 healthy age-matched control subjects were investigated, and VEGF, endothelial progenitor cell and immunoglobulins were not significantly different between the groups (P = 0.354). The endothelial progenitor cell count was inversely correlated with serum immunoglobulin G levels in the LCPD group (r = 0.403, P = 0.03). The absolute endothelial progenitor cell count was also significantly higher in the fragmentation stage than in the healing stage, and they were greater in bilaterally affected cases than in unilaterally affected patients. Similar results were found in a pig model[82]and Sprague-Dawley rats[81]. In the rat model, however, the interruption of vascularization in the proximal femoral growth plate was not followed by diffuse damage.
Insulin growth factor 1 (IGF-1):The role of IGF-1 role in LCPD aetiology was investigated. In particular, a study in spontaneously hypertensive rat (SHR)demonstrated altered IGF-I expression during early postnatal life and suggested that the altered IGF-I expression may cause the mechanical vulnerability of the femoral epiphysis. Low levels of serum IGF-1 and IGF-1 binding protein 3 have been reported in patients with LCPD[83,84]. However, these results conflict with another two studies that reported normal IGF-1 binding protein levels[85,86].
DISCUSSION
The aetiology of LCPD is still mainly unknown. The heterogeneous prevalence reported opens the discussion for the examination of possible environmental and social factors involved in the aetiology of the disease[1-3,29]. In support of this, three studies[21,26,87]reported a possible association between the decrease in the incidence and lifestyle changes over recent years, exposure to environmental risk factors like smoking, delayed epiphyseal ossification, low birthweight, child deprivation and obesity. However, the results are not definitive.
Several studies reported strong evidence supporting the role of social and economic deprivation in the incidence of LCPD in children[14,17,28,18-22,24-26]. This was proposed as a possible consequence of two factors: low birth weight and smoking habits. In addition, maternal smoking is associated with a low birthweight[88,89]. Thus, low birthweight could be often falsely associated with LCPD because of the smoking habit typically present in these families, which is the more probable cause of the higher incidence of LCPD. For these reasons, the role of low birth weight lacks strong evidence[12-17,29], and the smoking habit, being more common in social-economical deprived families, may play a more important role in the etiopathogenesis of LCPD.This was further confirmed by studies investigating the association between smoking and LCPD, which reported a significant augmentation of the risk in smokers and smoke-exposed family. The mechanism proposed is the smoke-dependent damage of vessel endothelium, which could ultimately lead to epiphyseal infarction[17].
In addition, recent evidence supports obesity as a major risk factor[38]. Leptin is a hormone that is expressed in adipose tissue and, at lower levels, in gastric epithelium and placenta associated with both obesity and bone metabolism[90,91]. It has been reported that both leptin and obesity are positively associated with the severity of LCPD[38,39]. Most obese humans have very high plasma leptin concentrations,suggesting they are resistant to its anorectic and metabolic effects[92]. Based on these findings, Bartell et al[93]conducted a study involving intracerebroventricular and subcutaneous administration of leptin in leptin-deficient ob/ob mice, investigating its effect on bone and muscular tissues. In both experiments, leptin had a key role in the decrease of body weight, food intake and body fat and in the increase of muscle mass,bone mineral density, bone mineral content, bone area, marrow adipocyte number and mineral apposition rate. Thus, leptin administration seemed to be really effective in obese patient bone metabolism. Following this rationale, Zhou et al[94]in 2015 tested these effects in a LCPD obese rat model. Six weeks after surgery induced avascular necrosis of the femoral head, radiologic and histomorphometric assessments were performed. Radiographs showed better preservation of the femoral head architecture in the leptin-treated group. Histology and immunohistochemistry revealed that the leptin group had significantly increased osteoblastic proliferation and vascularity in infarcted femoral heads compared with control groups. The mechanism proposed is linked to both a direct action of leptin on bone metabolism and an indirect action through the upregulation of VEGF[95]. In particular, leptin acts physiologically through MAPK/ERK 1/2 and PI-3K/AKT1 pathways and a series of transcription factors such as HIF-1α[92,96]. Emerging evidence supports how leptin and obesity may play a role in LCPD etiopathogenesis. We strongly encourage further studies on leptin and obesity,associated with their effects of bone metabolism, in LCPD patients. These studies should be conducted both as a therapeutic option and as a possible actor in the aetiology of the disease.
The dissimilarity in incidence between male and female subjects was initially thought to rely on the different etiopathogenesis of the disease. In particular, pioneer studies on these differences reported a more severe presentation and prognosis in female patients than in male subjects[97]. However, recent high profile epidemiological studies reported no significant differences in clinical presentation, outcome and prognosis between boys and girls[98,99]. Thus, the LCPD male/female ratio appears not to be associated with different clinical presentation and should not be part of the clinical treatment algorithm.
While the reviewed literature provided evidence of cases of LCPD running in families[41-46], and epidemiological studies suggested a genetic role[29], twin studies[27,28]reported no significant concordance. This was further investigated by another recent study[48]involving 37 patients and 100 controls that reported no difference in genotype variants and expression of coagulation and inflammatory factors. However, Zheng et al[100]investigated the relationship between global DNA methylation involving 82 children with LCPD and 120 matched controls. They reported significant differences in the global methylation of peripheral blood DNA between patients with LCPD and matched controls. Further high-profile epigenetic studies in key tissues should be conducted providing new protagonists of the LCPD aetiology. COL2A1 alterations were also investigated. Several case reports and studies were reported in our review[42-46]. Their association with the disease was found to be weak in a case study published by Kenet et al[101]involving 119 LCPD affected children and 276 controls,further supported by a review of the literature. In the same study, the absence of significant association with Gaucher's disease and Factor V Leiden alterations was also reported. On the contrary, further histological and ultrastructural studies found how p.Gly1170Ser mutation of COL2A1 is involved in the pathogenesis of a type II collagenopathy[55]. This alteration leads to an amino acid change that perturbs a Gly-XY triple-helix repeat, which is a fundamental structure in type II collagen function[102].Thus, COL2A1 alterations should be further studied to clarify their role in the pathogenesis and aetiology of the LCPD.
Besides factor V Leiden and Prothrombin II polymorphisms being associated with a three times higher likelihood of disease and a 1.5-fold increase of risk, the results were not statistically significant[47]. Thus, even though the topic was extensively studied,recent evidence of hypercoagulability is inconclusive. Baltzer et al[103]reported a case of a child affected by Kienbock’s disease and factor V thrombophilia who developed LCPD. The patient’s hypercoagulability state may link with the bilateral manifestation of Perthes disease. After a further review of the literature, the authors also found two other cases of multifocal osteonecrosis and hypercoagulable disorder in adult patients. We encourage additional molecular and clinical studies on hypercoagulative states and LCPD.
Among the studied molecules, inflammatory cytokines involved in immune responses provide a crucial prospective of research. In particular, a study[51]found how a polymorphism of IL-6, a proinflammatory cytokine involved in chronic inflammation and immune response[104], was found to be associated with LCPD. In addition, another study[52]reported a high synovial level of IL-6 in LCPD patients.This suggests that IL-6 is a possible future protagonist of new therapeutic approaches and translational research. We strongly encourage further studies on the IL-6 pathway. Another molecule studied is SIRT1, the mammalian ortholog of the yeast SIR2 (Silencing Information Regulator) and a member of the Sirtuin family. It was reported that it can inhibit nuclear factor kappa B (NF-kB) transactivational activity by deacetylating Lys310 of the RelA/p65 subunits. Thus, SIRT1 inhibits both its transcriptional activity and the release of inflammatory cytokines mediated by NF-kB[105]. What emerged in a recent research project held in 2017 is that treatment with interferon-β increased SIRT1 expression and inhibited secretion of IL-6 in avascular femoral head necrosis mouse model. Interferon-β activated SIRT1 in the RAW 264.7 cell and bone marrow-derived osteoclasts and decreased IL-6 secretion. What we can assume from these papers is that IL-6, NF-kB, SIRT1 and other molecules involved in the autoimmune response could be linked with LCPD aetiology and should be further investigated both as a possible cause of the disease and as a new potential therapeutic target.
A study of 2003[78]found how retarded epiphyseal ossification could be linked to LCPD. However, there is lack of consensus on the role of these anatomical alterations[65,79,80]. During development, the epiphyseal blood supply is almost exclusively provided by the deep branch of the medial femoral circumflex artery[75].Both imaging and histological studies have shown a partial or complete loss of blood flow[61,77]and the development of ischemic necrosis[54]of the femoral head in LCPD.Mechanical-induced ischemia is one of the most supported hypotheses for the aetiology of LCPD[54,61,75-77]. Emerging evidence is supporting a role of repetitive mechanical stress to the epiphysis and de subsequent ischemia, as stated in different experimental models[65-69,73,74]. In addition, the association with ADHD[30-34,36]and hyperactive modification of behaviour could be easily explained with repetitive activity-induced mechanical stress. This was further explained with an higher risk of injury[35]in LCPD patients. The recent evidence strongly supports the role of mechanical stress in the aetiology of LCPD. We encourage high profile clinical studies to investigate more this etiopathogenesis.
Our study has some strengths. We extensively searched and identified all relevant genetic association studies, the most common comorbid and the possible etiologic hypothesis. Therefore, risk of bias assessment showed moderate overall risk that could influence our analysis.
The literature available on the aetiology of LCPD presents major limitations in terms of great heterogeneity and lack of high-profile studies. Although a lot of studies focused on the genetic, biomechanical and radiological background of the disease,there is lack of consensus on one or multiple major actors of the etiopathogenesis.While obesity, smoking exposure and child deprivation seems to be associated with LCPD aetiology, more studies are needed to understand the complex and multifactorial genesis of the avascular necrosis characterizing the disease.
ARTICLE HIGHLIGHTS
Research background
Legg-Calvé-Perthes disease (LCPD) is a complex disease with a multifactorial aetiology. The etiopathogenesis of the disease was widely investigated in the last 20 years, but it is still unknown.
Research motivation
Numerous studies tried to explain the major actors in LCPD aetiology, but there is a lack of synthesis of the evidence.
Research objectives
The purpose of the study was to summarize the current evidence on the aetiology of LCPD.
Research methods
Two databases (PubMed and Science Direct) were systematically searched for relevant articles by two independent reviewers from their date of inception to the 20th of May 2018. Every step of the review was done according to PRISMA guidelines. Due to article heterogeneity and the topic after data analysis, a descriptive (synthesis) analysis was performed.
Research results
Sixty-four articles were included in this systematic review after applying our inclusion and exclusion criteria. Available evidence on LCPD aetiology is still inconclusive. Several hypotheses have been researched but none of them was found decisive.
Research conclusions
After our systematic review of the available evidence we conclude that LCPD aetiology relies on a multifactorial basis where environment in genetically predisposed patients participates in the pathogenesis of the disease.
Research perspectives
Further clinical and preclinical studies are strongly encouraged to understand better the mechanical and vascular basis of the etiopathogenesis of the disease. Interesting perspectives from studies on Leptin, obesity, and mechanical trauma were found and should be further investigated.
杂志排行

World Journal of Orthopedics的其它文章
- Linkage of microbiota and osteoporosis: A mini literature review
- Short-term differences in anterior knee pain and clinical outcomes between rotating and fixed platform posterior stabilized total knee arthroplasty with a new femoral component design
- Comparison of implant related complications amongst patients with opioid use disorder and non-users following total knee arthroplasty
- Changing trends in the mortality rate at 1-year post hip fracture - a systematic review