基于关键词的蛋白质交互关系识别
2019-03-21毛宇薇
毛宇薇,牛 耘
(南京航空航天大学 计算机科学与技术学院,江苏 南京 211106)
0 引 言
蛋白质是生物细胞的重要组成成分,细胞中多数重要的分子过程均通过蛋白质交互作用完成。蛋白质交互关系(protein-protein interaction,PPI)的识别对于生命过程的研究、疾病的治疗以及新药的研制具有十分重要的意义,是解决大量医学难题的关键信息。随着近年来互联网的迅速发展,生物医学文献数量飞速增长,以人工识别的方式从文献中获取蛋白质交互关系已经难以满足实际应用的需求。因此,利用信息抽取技术自动识别PPI已经成为一项重要的研究内容。
目前用于PPI识别的技术主要分为以下几种:基于共现的方法[1]、基于模式匹配的方法[2]、基于自然语言处理的方法[3-4]和基于机器学习的方法[5-6]。基于共现的方法通过统计蛋白质对在句子中出现的概率来识别蛋白质之间是否存在交互关系,这种方法召回率较高,但缺点在于无法识别出词典外的PPI。基于模式匹配的方法通过事先建立的模式集合在文本中进行匹配来识别PPI,可以获得较高的精确度,但是人工构造模式集合需要耗费大量的人力物力。基于机器学习的方法通过将PPI识别转化为文本分类问题,同时结合自然语言处理技术,可以很好地利用已有的大量文本数据,是目前PPI识别的主要研究方法[7-8]。这种方法首先通过自然语言处理技术对文本进行处理并抽取句子中的语言学特征对蛋白质交互关系进行表示,然后利用机器学习方法从大规模文本数据中学习并建立模型来对未知蛋白质交互关系进行识别。传统的机器学习方法根据语料标注情况可以分为有监督和无监督的方法。有监督的方法利用大量有标注的训练语料来训练分类模型,无监督的方法不依赖于标注语料,可以自动识别文本中的PPI[9-10]。有监督的方法对于标注语料的要求较高,而无监督的聚类方法选择缺少理论指导,二者均难以满足实际的应用需求,因而出现了基于弱监督的PPI识别方法[9]。
文中采用的基于弱监督的方法结合了有监督方法和无监督方法的优点,通过扩充少量有标注的种子集来对PPI进行识别,既节省了人工标注语料的成本,同时具有较好的性能。
1 基础PPI识别框架
采用的蛋白质交互关系识别框架基于弱监督的方法,仅需少量有标注的语料进行PPI识别。首先,针对语料中的每一个句子,提取能够表达蛋白质交互关系的特征作为词汇模式,以语料中少量带标注的蛋白质对作为种子集,依照分布式假设原理,根据词汇模式在种子集中的分布对其进行向量化表示。然后,通过相似性计算,将语料中与种子词汇模式相似的词汇模式挑选出,视为表达交互关系并加入候选集中;最后对候选集中的词汇模式进行打分,根据词汇模式的分数计算相应蛋白质对的得分,将得分大于阈值的蛋白质对加入种子集中。不断重复该过程,通过迭代完成蛋白质交互关系的识别。
1.1 向量空间模型构建
对每一个目标蛋白质对(protein1,protein2),检索出数据库中同时包含protein1和protein2的句子集合,作为该蛋白质对的签名档。通过观察发现,句子中两个蛋白质之间的动词和名词在该蛋白质对交互关系的表达上起到了重要作用。因此,提取两个蛋白质之间的动词和名词,去掉无实际意义的停止词,作为表达蛋白质交互关系的词汇模式。
分布式假设原理为相似上下文的单词具有相似的语义。根据该假设,如果两个句子的词汇模式出现在相似的蛋白质对中,则这两个词汇模式具有相似的语义关系。根据词汇模式在种子集中的分布对其进行向量表示,向量的维度即为种子集中蛋白质对的个数,由此得到向量空间模型。
1.2 聚类词汇模式并产生候选集
以少量有交互关系的蛋白质对作为初始种子集,对种子蛋白质对的词汇模式采用序列聚类算法,将在种子集中分布相似的词汇模式聚为簇,得到语义关系相似的模式簇。每一个簇中的词汇模式都分布在相似的蛋白质对中,因此具有相似的语义关系。
在序列聚类算法中,聚类前对词汇模式的排序极为重要,对聚类结果有较大影响。在基础的算法框架中,根据词汇模式集所共现的种子蛋白质对个数进行降序排序,并且删除与种子蛋白质对共现次数过少的词汇模式。该排序方式依据的原理为:若词汇模式与越多的种子蛋白质对共现,则其表达交互关系的可能性越高。
聚类后,将语料库中的词汇模式与所得到的语义关系簇进行相似性计算,将相似度大于阈值Q的词汇模式及其所对应的蛋白质对加入候选集。根据每一个簇中挑选出的词汇模式计算该簇的得分,表示该簇挑选出的有交互词汇模式的可靠性。
1.3 评估候选集并更新种子集
对候选集中的每一个词汇模式,根据挑选出该模式的语义关系簇得分及模式与簇之间的相似度,可计算得出该词汇模式的分数。通过蛋白质对所对应的所有词汇模式的得分,可进一步计算得到蛋白质对得分。挑选出分数大于阈值T的蛋白质对作为本次迭代所识别出的有交互关系的蛋白质对,加入到种子集。不断迭代上述过程扩充种子集,直到满足终止条件,最终完成对蛋白质交互关系的识别。
2 基于关键词的改进PPI识别
基础的蛋白质交互识别框架仅以词汇模式与种子蛋白质对的共现次数为线索对模式进行排序与挑选。虽然一部分与种子蛋白质对共现较多的词汇模式有可能表达交互关系,有些词汇模式,如:express(表达)等,虽与较多种子共现,但并不能表达交互关系。因此,这种方法可能将一些未表达交互关系的词汇模式当作较为可靠的表达交互关系的模式,排在模式集合前面,影响了聚类结果,进而影响最终的识别效果。
通过对蛋白质对签名档的观察,一些关键词,如:inhabit(抑制)、induce(引起)等能够较好地表达蛋白质对交互关系。因此,如果一个词汇模式包含有关键词,则认为该词汇模式更有可能表达交互关系;并且如果一个关键词越有可能表达交互关系,则包含有该关键词的词汇模式也更可能表达交互关系。文中提出利用关键词对聚类算法的排序过程进行改进,将包含可靠关键词的词汇模式排在模式集的头部,并且去掉没有关键词的词汇模式,改善聚类效果,使得语义关系簇所表达的交互关系更为准确。
2.1 基于词向量的关键词扩充
文中采用的初始关键词为Joshua M等[11]提出的表达蛋白质交互关系的关键词,共179个。这些关键词虽然能精确表达交互关系,但并不全面,有些不包含于这些关键词中的单词也可能表达交互关系。因此,根据初始关键词集,采用基于词向量的方法,挑选出与初始关键词相似度较高的单词作为新的关键词,并对新关键词表达交互关系的可靠性进行评分。
2.2 词向量
采用词向量(word embedding)来计算单词之间的相似度。词向量作为深度学习(deep learning)模型中一种单词的分布式表达(distributed representation),最早由Hinton提出[12-13]。相对于传统的独热表示(one-hot representation)方法,该表示方法最大的优点在于可以通过向量的距离衡量词之间的相似度;此外,通过低维表示的词向量大幅减小了计算的复杂度,从而提高了方法的实用程度[14-15]。
词向量通常利用无监督的语言模型在大量未标注的文本数据中训练得到,文中对蛋白质对签名档中的所有句子所形成的初始语料进行预处理,作为训练数据,具体预处理过程如下:
(1)在生物医学文献中存在大量种类繁多的生物体名称,这些存在于当前词上下文中的不同生物体名称对词向量的训练会有较大的影响。因此将不同种类的生物体名称按照其类型进行替换,如:将蛋白质名称“cd4”、“cd44”等替换为“Protein”。
(2)对句子进行分词处理,并去掉标点符号(除了连词符‘-’),去掉数字、单字符单词和无实际意义的停止词。
(3)对单词进行词干提取,使相同词干的单词变为同一个单词,如:induce,induced,induces都变为induc。
通过Skip-gram模型训练经过上述处理后所得到的训练数据,得到词向量模型,根据该模型可计算得到签名档中每个单词的词向量。
2.3 关键词生成
根据初始关键词集合K和2.1所得到的词向量,算法1描述了关键词生成算法,算法输出新的关键词集合,其中包含每个关键词及其表达交互关系的得分。
输入:初始关键词集合K={k1,k2,…,kn},词向量W={w1,w2,…,wn},阈值θ
输出:新关键词与对应分数集合N={[n1,s1],[n2,s2],…,[nn,sn]}
1:N={}
2:forki∈Kdo
3:forwi∈Wdo
4:sim(ki=sim(ki)∪calc_sim(ki,wi,θ))
5:end for
6:sort(sim(ki))
7:for [wi,simi]∈sim(ki) do
8:mark=0
9:for [ni,si]∈Ndo
10:ifwi==nithen
11:sj+=simi
12:mark=1
13:end if
14:end for
15:if mark==0 then
16:N=N∪[wi,simi]
17:end if
18:end for
19:end for
20:returnN
首先,初始化新关键词集合N。外层循环(第2步)遍历集合K中的每一个初始关键词ki,内层循环(第3步)遍历每一个单词的词向量wi,函数calc_sim(ki,wi,θ)计算ki和wi的相似度,返回相似度simi。如果相似度simi大于阈值θ,则把wi及相似度simi加入到集合sim(ki)中。函数sort(sim(ki))对集合sim(ki)按相似度降序排序。接着,外层循环(第7步)遍历集合sim(ki)中的每一个关键词,标志mark初始化为0,表示该关键词不在新关键词集合N中。内层循环(第9步)遍历新关键词集合N,如果wi等于nj,则表示该关键词已存在于集合N中,将原先的分数sj加上相似度simi得到新的分数,并将标志mark赋值为1。如果内层循环结束,标志mark等于0,则表明该关键词wi不在集合N中,将相似度simi作为wi的分数,加入到集合N中。最后,函数Norm(N)将所有的关键词分数除以最大的分数,更新每个关键词的得分,返回关键词集合N。
2.4 基于关键词的改进算法
基于文中语料通过2.1可以得到能够表达交互关系的新关键词及其表达交互关系的可靠性分数。将初始关键词的分数设置为1,与新关键词合并,得到关键词集合。根据该关键词集合对聚类算法做如下改进:
(1)遍历输入的种子词汇模式集合,对每一个词汇模式,如果其中含有关键词,则认为该词汇模式较可能表达交互关系,予以保留;否则认为该模式表达交互关系的可能性过低,去掉该词汇模式。
(2)对集合进行重新排序:首先,根据每一个词汇模式中所包含的关键词的分数降序排序,如果一个词汇模式中包含有两个及以上的关键词,则根据分数较大的关键词进行排序;然后,对于所含关键词分数一样的词汇模式,根据其与种子集共现的次数进行降序排序。最终得到的词汇模式集以关键词的分数为首要排序依据,排在集合头部的词汇模式含有分数较高的关键词,其表达交互关系的可能性更大。
3 实 验
3.1 实验数据及设置
实验中采用的有交互关系蛋白质对来源于专业PPI数据库HPRD[14],并且只保留出现在PubMed数据库[15]一篇以上摘要里的蛋白质对。同时,采用生物医学领域的常用方法,将获得的有交互蛋白质对进行随机组合,去除其中已经包含在HPRD中的蛋白质对。最终分别得到1 141对有交互关系的蛋白质对和1 353对无交互关系的蛋白质对。对每一个蛋白质对,检索PubMed数据库的文献摘要,并提取所有包含此蛋白质对的句子构成该蛋白质对的签名档。所有的2 494个蛋白质对及其签名档构成文中的语料库,并从有交互关系的蛋白质对中随机选出100对构成种子集。
实验过程中所需要设置的参数有两个:一是挑选与语义关系簇相似度大于阈值Q的词汇模式加入候选集中,阈值Q设置为0.6;二是选择分数大于阈值θ的关键词为新增加的表达交互关系的关键词,这里的阈值θ设置为0.7。实验采用的结果性能评价指标是当前PPI抽取系统主要使用的3个指标:精确度、召回率和F值。
Precision=TP/(TP+FP)
Recall=TP/(TP+FN)
F-Score=2×P×R/(P+R)
3.2 实验结果及分析
表1为基础PPI识别框架第一次迭代后,阈值T的不同取值所得到的结果。
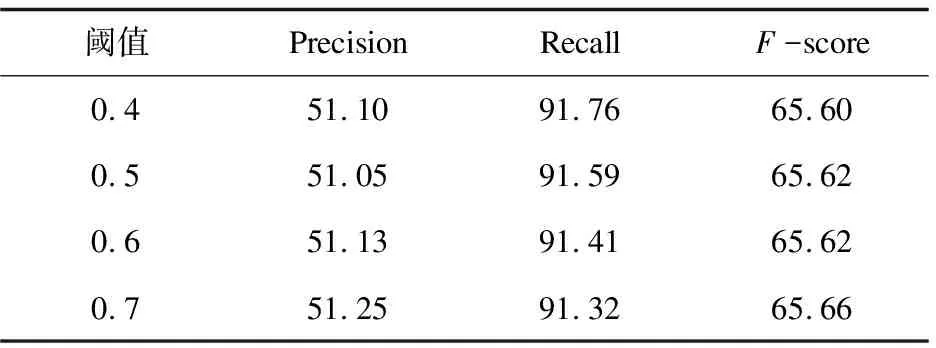
表1 基础框架不同阈值结果对比 %
从表1可以看出,随着T值的增大,识别的精确度上升,召回率下降,总体F值上升。阈值为0.4时,得到的召回率最高为91.76%,精确度最低为51.05%,总体F值也最低,为65.60%。阈值为0.7时,得到的召回率最低为91.32%,精确度最高为51.23%,总体F值也最高,为65.66%。0.4到0.7的所有阈值T取值所得的4个结果,虽然召回率都较高,但精确度都较低,精确度与召回率的值差距较大,因此总体F值都不高。
表2为添加了关键词模块的改进PPI识别算法,通过第一次迭代过程,根据阈值T的不同取值所得到的结果。
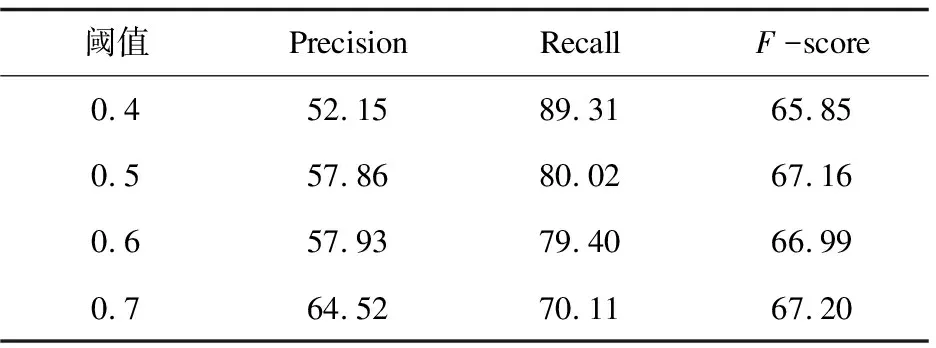
表2 改进算法不同阈值结果对比 %
从表2可以看出,随着T值从0.4增大到0.5,识别的精确度从52.15%显著增加到57.86%,召回率下降,总体F值上升。T值从0.5增加到0.6的结果影响不大。当T值从0.6增大到0.7时,精确度从57.93%显著增加到64.52%,召回率下降,总体F值上升。T值为0.7时F值最高,为67.20%。
对比表1和表2可以发现,在考虑了关键词这一线索后,识别的精确度有了显著提高,虽然召回率下降,但整体F值有明显提高。其中当阈值为0.7时,两种算法都达到了最高的F值,相较于改进前的基础框架,改进后的算法的精确度提高了13.27%,召回率下降了21.21%,总体F值提高了1.54%。由实验结果可知,通过关键词对种子词汇模式集进行挑选,以及在聚类前对模式集进行排序,能够明显提高聚类的效果,使得语义关系簇中的词汇模式表达交互关系的可靠性更高,进而后续对语料词汇模式的识别更加准确,使得蛋白质对识别的精确度明显提高。因为将没有关键词的词汇模式删除,使得有些可能表达交互关系但没有关键词的词汇模式被去掉,导致召回率下降。实验结果表明总体F值有明显提升,该基于关键词的改进算法对PPI识别有着较好的改进效果。同时,通过该步骤,聚类算法的输入词汇模式数从原先的8 520个减少到3 493个(仅为原先的41%),极大地提高了算法的效率,运算速度明显提高。
设置第一次迭代后F值取得最高时的阈值T=0.7作为迭代过程中挑选蛋白质对加入种子集的阈值。表3为改进算法的阈值T取0.7时,每一次迭代的实验结果。
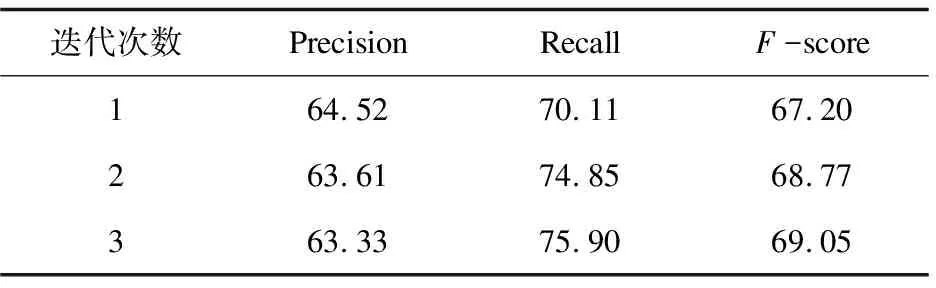
表3 迭代结果 %
从表3可以看出,随着迭代次数的增加,蛋白质对交互关系的识别精确度略有下降,召回率有较明显提升,整体F值上升,同时第3次迭代后的结果与第2次的迭代结果相差不大。该实验结果表明迭代过程可以较好地提高算法的性能,并且经过3次迭代后算法已经取得了较好的结果,3次迭代后的F值可达到69.05%。
4 结束语
文中的基础蛋白质交互识别算法框架采用弱监督的方法,仅需少量有交互关系的蛋白质对作为种子集,利用相似性聚类方法迭代扩充种子集,最终完成交互关系的识别。利用关键词能够较好地表达蛋白质交互关系这一线索,该改进算法基于词向量对初始关键词进行扩充,并根据扩充后的关键词集合对聚类算法的输入词汇模式集合进行挑选与排序,提高聚类算法的效果进而提高最终的识别结果。实验结果表明,该方法以较少的有标注数据取得了较高的精确度与召回率,同时改进后的算法与之前的基础算法相比,精确度明显提升,总体F值也有一定提高。
目前的方法仅在聚类算法中利用关键词这一线索,之后的研究将进一步在算法的其他步骤,如候选集的评分机制中考虑关键词这一线索,并且将挖掘其他在表达交互关系中较为重要的线索。
