小规格药物粉末混合均匀性评价取样方式研究*
2019-03-06郑华樟张均平罗丽娟朱文涛欧阳婷
郑华樟,张均平,罗丽娟,朱文涛,欧阳婷
(江西青峰药业有限公司药物研究中心, 江西 赣州 341000)
小规格药物是指每一个单剂标示量小于25 mg或主药含量小于每一个单剂重量25%[1]。物料充分混合均匀是保证固体制剂质量均一性和稳定性的关键,因此小规格药物粉末混合工艺是固体制剂生产的关键工序。评价药物粉末是否混合均匀是药品生产过程中一项重要的中间产品控制策略,而评价药物粉末混合均匀性的取样方式是否合理,对于评价药物粉末混合均匀性尤为重要[2-7]。
2003年11月 FDA发布了《粉末混合和单位成品制剂分层取样和评估的工业指导原则》[8],该指导原则描述了药物粉末混合均匀性评价取样方式。2010版《口服固体制剂GMP实施指南》[9]同样对药物粉末混合均匀性评价取样方式进行了阐述。参考上述指导原则,本文比较了不同的取样方式对小规格药物粉末混合均匀性评价的影响。
药物A含有多个酰胺结构,在酸性、碱性条件可发生降解。药物A为结晶性粉末,存在引湿性,粒径较小d(0.9)<50 μm,有一定粘附性。
1 材料与方法
1.1仪器与试剂SYH-10三维运动混合机(常州市震华干燥设备有限公司),ZP14旋转式压片机(北京国药龙立科技有限公司),LE204E/02分析天平(瑞士梅特勒-托利多集团股份有限公司),SHZ-B水浴恒温振荡器(上海博讯实业有限公司),安捷伦1260高效液相色谱仪(安捷伦科技有限公司)。
分析级磷酸二氢钾(国药集团药业股份有限公司),分析级磷酸(西陇化工股份有限公司),色谱级乙腈(TEDIA有限公司)。
1.2制备工艺药物A为我公司开发的小规格品种,规格为0.5 mg,单位剂量片重为100 mg,处方中填充剂为3∶1比例的微晶纤维素和预胶化淀粉,润滑剂为0.5 mg/片的硬脂酸镁。原料药在处方中占比极小,因此采取等量递增的混合方式对原辅料进行混合。将原料药、辅料分次备量加入三维运动混合机混合,设定频率10 Hz,每次混合10 min。原辅料等量递增混合结束后加入硬脂酸镁进行总混,总混结束后按照理论片重100 mg进行压片。
1.3取样按照上述工艺参数混合,总混结束后用单点取样器在三维运动混合机混合筒内多个不同部位取样,将取出的样品按照规定量称量后倒入容量瓶或自封袋内。取样位置示意图如图1。
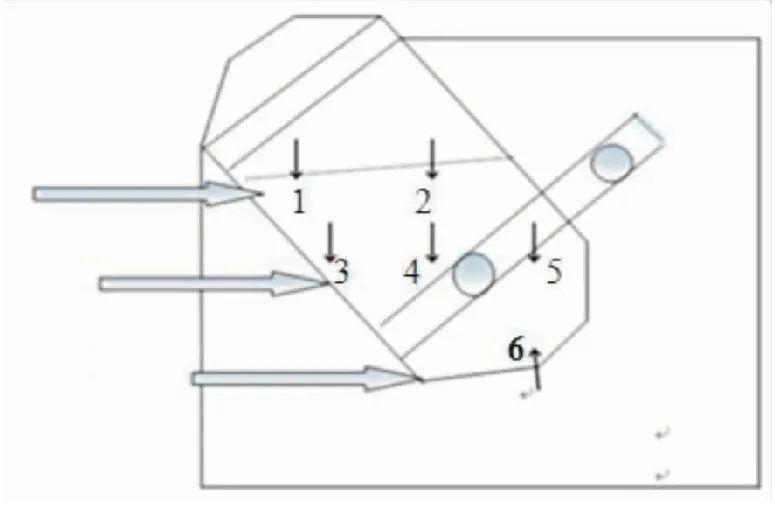
图1 药物混合粉末混合均匀度取样示意图
1.3.1取样方式1 按照图1取样示意图在三维运动混合机6个不同部位取样。用装有0.5 mL取样器头的单点取样器在每个取样位置取样,取出的样品用分析天平精密称量180 mg±30%(126~234 mg),每份样品称样量见表1,全量转移至50 mL容量瓶中。
1.3.2取样方式2 按照图1取样示意图在三维运动混合机6个不同部位取样。用装有1.5 mL取样器头的单点取样器在每个取样位置取样,取出的样品称量后装入自封袋内,取样量见表2。
1.4药物粉末含量均匀度测定
1.4.1色谱条件用十八烷基硅烷键合硅胶为填充剂;以0.03 mol·L-1磷酸二氢钾溶液(用磷酸调节pH值至3.0)-乙腈(83︰17)为流动相;柱温30 ℃;流速每分钟1.0 m;检测波长为342 nm。
1.4.2供试液配制分别往每个装有样品的50 mL容量瓶中加水适量,置于恒温水浴振荡器内运行30 min,加水稀释至刻度,摇匀,滤过即可。两种取样方式的样品配制量分别见表1、表2。
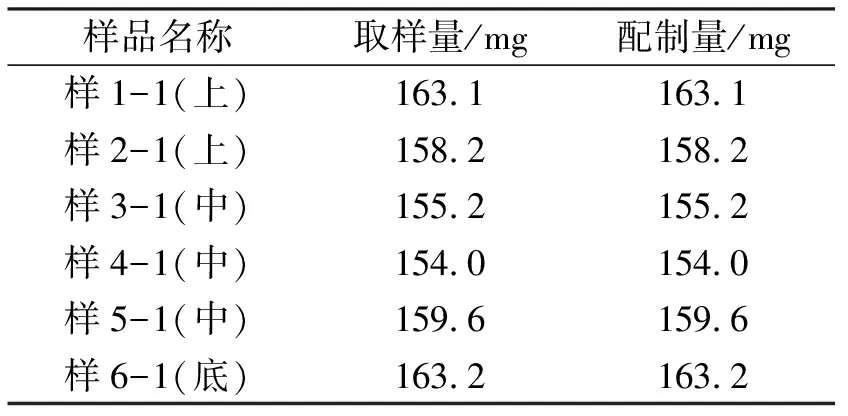
表1 取样方式1样品取样量及配制量
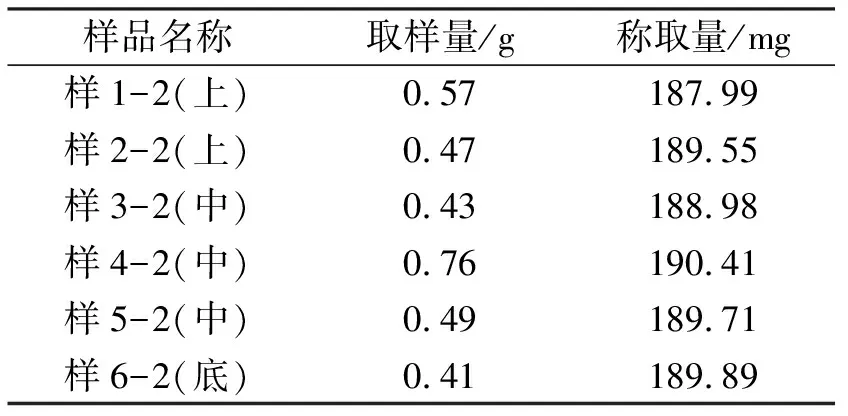
表2 取样方式2样品取样量及称取量
1.4.3线性与范围取药物A对照品适量,配制成相对供试品溶液浓度50%(10.237 7 μg·mL-1)、70%(14.332 8 μg·mL-1)、80%(16.380 3 μg·mL-1)、100%(20.475 4 μg·mL-1)、120%(24.570 4 μg·mL-1)、130%(26.618 0 μg·mL-1)、150%(30.713 0 μg·mL-1)的溶液,依法检测,以溶液组分的峰面积A对相应的溶液浓度C进行线性回归,绘制标准曲线,结果表明药物A在供试品浓度的50%~150%的范围内,药物A的浓度与峰面积的线性关系良好,结果见图2。
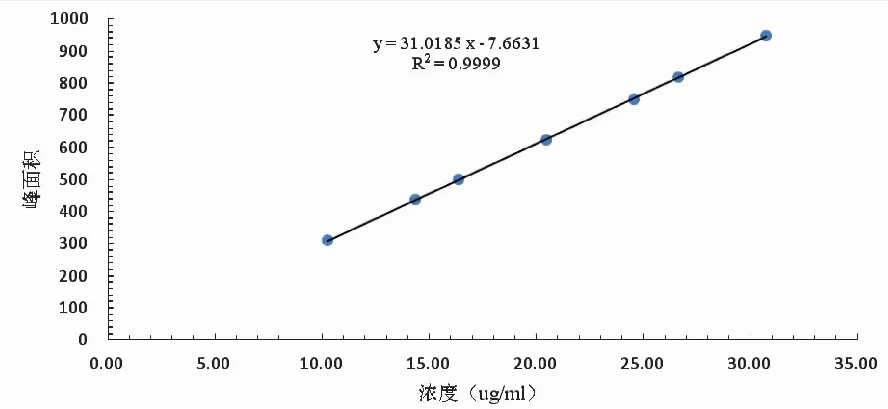
图2 含量及含量均匀度线性曲线图
1.4.4准确度和精密度取药物A原料药适量,加处方量空白辅料,配制成进样浓度50%(3份)、100%(6份)、150%(3份)的样品,依法进样检测,计算回收率,以此验证准确度。其中100%(6份)的回收率测定结果作为重复性实验;同法,由不同实验人员,不同日期,使用不同的色谱系统,配制100%(6份)样品,作为中间精密度实验。
3个测试浓度(相对供试品溶液浓度50%、100%、150%)的平均回收率分别为99.5%、99.5%、99.3%,综合回收率为99.4%,12份测试样品的回收率测试结果相对标准偏差为0.5%,表明准确度良好。重复性实验和中间精密度实验的平均回收率分别为99.5%、100.2%;回收率的相对标准偏差分别为0.4%、0.2%;12份测试样品回收率测试结果相对标准偏差为0.5%,表明中间精密度和重复性良好。
1.4.5溶液稳定性考察依法配制对照品溶液、供试品溶液,分别于0 h、12 h、24 h、36 h、48 h、60 h、72 h进样,计算溶液主峰面积的相对标准偏差,考察溶液稳定性。测得对照品溶液峰面积RSD为0.21%,供试品溶液峰面积RSD为0.17%,表明对照品溶液和供试品溶液在室温条件下72 h溶液稳定。
1.4.6粉末含量均匀性测定分别对取样方式1和取样方式2获得的样品进行含量均匀度测定。按照含量测定的色谱条件与系统适用性试验,以磷酸二氢钾溶液(用磷酸调节pH至3.0)-乙腈(83︰17)为流动相;柱温30 ℃;流速每分钟1.0 mL。取对照品约10 mg,置50 mL量瓶中,加水溶解并稀释至刻度,取50 μL注入液相色谱仪,记录色谱图;精密量取供试续滤液50 μL注入液相色谱仪,同法测定记录色谱图;按外标法以峰面积计算每份样品含量及RSD值。
1.5压片将药物总混粉末进行压片,通过片剂含量均匀度结果验证药物粉末是否混合均匀,最终判定哪种取样方式能够正确评价药物粉末混合均匀度。
使用ZP14旋转式压片机进行压片,应压片重100 mg,片重范围控制±5%(95~105 mg),在压片过程的前段(正式压片开始后5分钟内)、中段(压片中间过程)、后段(压片结束前5分钟内)分别取样检测片剂含量均匀度。
2 结 果
2.1取样方式1、取样方式2药物粉末含量测定结果表3显示,取样方式1总混药物粉末平均含量0.58%,含量RSD 2.3%,显示药物粉末混合均匀。表4显示,取样方式2总混粉末平均含量0.44%,超出理论含量标准范围;含量RSD为10.3%,大于5.0%,显示药物粉末未混合均匀。
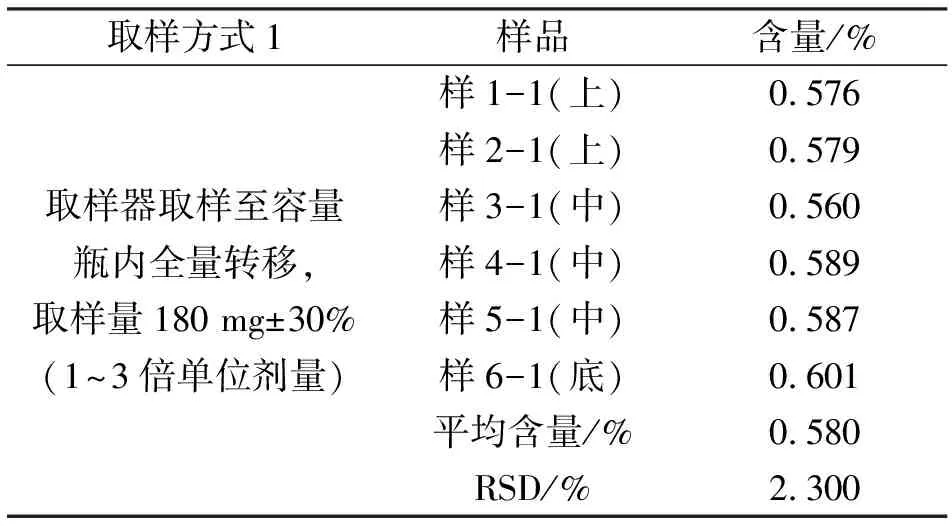
表3 取样方式1样品含量测定结果
注:理论含量为0.555%,含量标准范围(0.499%~0.611%),RSD≤5.0%,证明药物粉末混合均匀。样品测定次数n=3。

表4 取样方式2样品含量测定结果
注:理论含量为0.555%,含量标准范围(0.499%~0.611%),RSD>5.0%,证明药物粉末未混合均匀。样品测定次数n=3。
2.2压片过程中片剂含量均匀度测定片剂压片过程中前中后段含量均在99%以上,A+2.2S<15.0,片剂含量及含量均匀度合格。见表5。

表5 压片过程中片剂含量均匀度测定结果
注:片剂含量标准范围90.0%~110.0%,“A”为标示含量与含量均值之差的绝对值。“S”为含量标准差。
3 讨 论
表3总混药物粉末平均含量0.58%,含量RSD 2.3%,显示药物粉末混合均匀。表4总混粉末平均含量0.44%,超出理论含量标准范围;含量RSD为10.3%,大于5.0%,显示药物粉末未混合均匀。表5数据显示片剂在压片过程中含量均匀度合格,证明药物粉末混合均匀。由此可确定取样方式1:将药物粉末取至容量瓶内全量转移的取样方式合适可操作。
将样品取样至自封袋内再进行分样的方式带来的误差会使得本身混合均匀的样品出现不均匀的假象。有多种因素会造成这一结果, 例如取样器设计和操作、取样技术、静电荷以及配方中物料的物理性状[5]。尤其对于低剂量粘附性较大的药物混粉在取样转移过程中容易导致原料吸附在塑料袋上出现含量偏低。从自封袋内将药物进行称量分样的过程也可能使药物粉末发生分离或者脱混。由此可知取样方式对评价药物混合均匀性有较大影响,需要从取样量、转移方式等方面考察选择最合适的取样方式[10]。对于小规格药物粉末混合均匀性评价取样方式可综合参考FDA《粉末混合和单位成品制剂分层取样和评估的工业指导原则》、2010版《口服固体制剂GMP实施指南》、国际制药工程协会ISPE推荐的取样方法[11-13],同时结合药物本身的性质选用合适的取样器取样并将药物粉末全量转移,取样量一般为1~3倍药物单位剂量。
