Pharmacology and clinical applications of flupirtine: Current and future options
2019-02-21KimLawson
Kim Lawson
Abstract Flupirtine is the first representative in a class of triaminopyridines that exhibits pharmacological properties leading to the suppression of over-excitability of neuronal and non-neuronal cells. Consequently, this drug has been used as a centrally acting analgesic in patients with a range of acute and persistent pain conditions without the adverse effects characteristic of opioids and non-steroidal anti-inflammatory drug and is well tolerated. The pharmacological profile exhibited involves actions on several cellular targets, including Kv7 channels, G-protein-regulated inwardly rectifying K channels and γ-aminobutyric acid type A receptors, but also there is evidence of additional as yet unidentified mechanisms of action involved in the effects of flupirtine. Flupirtine has exhibited effects in a range of cells and tissues related to the locations of these targets. In additional to analgesia, flupirtine has demonstrated pharmacological properties consistent with use as an anticonvulsant, a neuroprotectant, skeletal and smooth muscle relaxant, in treatment of auditory and visual disorders, and treatment of memory and cognitive impairment. Flupirtine is providing important information and clues regarding novel mechanistic approaches to the treatment of a range of clinical conditions involving hyper-excitability of cells. Identification of molecules exhibiting specificity for the pharmacological targets (e.g., Kv7 isoforms) involved in the actions of flupirtine will provide further insight into clinical applications.Whether the broad-spectrum pharmacology of flupirtine or target-specific actions is preferential to gain benefit, especially in complex clinical conditions, requires further investigation. This review will consider recent advancement in understanding of the pharmacological profile and related clinical applications of flupirtine.
Key words: Flupirtine; Kv7 channels; GABAA receptors; Analgesia; Seizures;Neuroprotection; Myotonia; Memory; Tinnitus
INTRODUCTION
Flupirtine is the first representative in a class of triaminopyridines that exhibits pharmacological properties leading to the suppression of neuronal over-excitability.Consequently, this molecule has demonstrated to be beneficial in treating patients with a range of pain conditions[1-4]. Flupirtine relative to other analgesics on the market exhibits unique chemical structure and modes of action that contribute to a preferable pharmacological profile. It does not possess the adverse effects characteristic of opioids and non-steroidal anti-inflammatory drug and is well tolerated.
Flupirtine has been classified as a selective neuronal potassium channel opener due to action on voltage-gated K channels belonging to the Kv7 subfamily (with selectivity for the Kv7.2-Kv7.5 isoforms) and G-protein-regulated inwardly rectifying K (GIRK)channels[5,6]. Channel activation by flupirtine will lead to hyperpolarization of the membrane potential and attenuates the generation of action potentials, thus offering a novel therapeutic approach for diseases associated with cellular hyperexcitability[5].
Flupirtine has been the subject of a few good reviews describing it’s history and clinical profile as a treatment of pain[1-4]. In addition, flupirtine has been used as a pharmacological tool to gain greater understanding of Kv7 channels as a therapeutic target[7-9]. The aim of this review is to consider the advancement in recent understanding of the pharmacological profile and related clinical applications of flupirtine. MEDLINE database, Web of Science and Google Scholar were used to identify relevant studies and publications up to July 2018 using the term “flupirtine”.Flupirtine exhibits an interesting pharmacological profile that offers clues of potential targets that merit investigation as novel therapeutic approaches to a range of clinical conditions.
ANALGESIA
Pain relieving activity in various animal models and humans has been demonstrated with flupirtine a non-opioid analgesic without anti-inflammatory or antipyretic properties[1-3]. Effective analgesia by flupirtine has been demonstrated in a range of persistent pain conditions such as musculoskeletal pain, postoperative pain, migraine and neuralgia[2,3,10,11]. These effects of flupirtine are associated with restoration of normal sensitivity of over-excitable nociceptive pathways and inhibition of the stimulation of nociceptive neurons by factors such as inflammatory mediators (e.g.,bradykinin)[12-15]. In small fibre neuropathy the efficacy of flupirtine was reported to be sufficient to lead to the discontinuation of first-line drug treatments, such as gabapentin and amitriptyline, which are often associated with adverse effects or unsatisfactory pain relief[16].
Flupirtine stabilizes the membrane resting potential by activating KCNQ (Kv7)potassium channels generating a neuronal hyperpolarizing current (M-current)[5].Activation of potassium channels will lead to an indirect N-methyl-D-aspartate(NMDA) receptor antagonism, thus reducing hyperexcitation of nociceptive neurons.In addition, Mg2+block on NMDA receptors is maintained by an oxidizing action of flupirtine at the redox site of the receptor consistent with an indirect inhibition[17].There is however no interaction with the binding site of the NMDA receptor[6].
Kv7 channels encompass five members of which Kv7.2–Kv7.5 are expressed and distributed throughout peripheral nerves and the CNS, and co-assemble to form either homo- or hetero-tetramers[18,19]. Agonist action at (Gq/11) G protein-coupled receptors, such as acetylcholine at muscarinic receptors, can inhibit channel activity.Kv7 channels are expressed in neurons forming the nociceptive pathways, such as central terminals of primary afferents and dorsal horn neurons within the spinal cord[20]. Long-term downregulation of Kv7 subunits following neuropathic injury,whether due to trauma or inflammation, has been proposed to contribute to hyperexcitability of sensory fibres[21]. Increased levels of repressor element1-silencing transcription factor leading to reduced density of Kv7.2, Kv7.3 and Kv7.5 is a delayed feature of neuropathic injury with probable involvement in the maintenance rather than the initiation of pain[22]. The decreased channel density was functionally compensated by the activation of residual Kv7 channels by flupirtine resetting neurons to a low-excitable state and suppressing pain[21]. Flupirtine exhibits efficacy on all four subunits, Kv7.2, Kv7.3, Kv7.4 and Kv7.5, with an apparent preference for Kv7.3. Interestingly, flupirtine appeared to evoke an effect specifically in injured neurons and not uninjured fibres[21]. Associated with the analgesic activity of flupirtine is the ability of inhibiting the release of neurotransmitters, such as calcitonin-gene related peptide, from the brainstem following the opening of Kv7 channels[23].
Flupirtine has been found to act simultaneously on Kv7 channels and γaminobutyric acid type A (GABAA) receptors which are both involved in the control of nociception[24]. Thus, the combined stimulatory action in pain neural circuits may contribute to the analgesic activity of flupirtine. The subunit composition of the GABA receptor defines specific pharmacological characteristics such that benzodiazepines modulate γ2-subunit containing receptors, whereas δ-subunits are highly sensitive towards neurosteroids[25,26]. Preferential action by flupirtine at δsubunit containing GABAAreceptors over γ-containing GABAAreceptors has been demonstrated[27]. The presence of α4 and α6 subunits renders δ-subunit containing GABAAreceptors insensitive to benzodiazepines[28,29]. Thus, the pharmacological properties of flupirtine due to interaction with GABA receptors will have a profile that differs from that exhibited by benzodiazepines. Although the induction of addictive behaviours in certain drugs is associated with agonism at GABAAreceptors,such properties of flupirtine have had limited anecdotal reporting[30,31].
Synergistic or additive effects with other analgesics have been suggested to be likely for molecules that inhibit the NMDA receptor[32]. Flupirtine and opioids have been reported to exhibit synergistic analgesic interactions[32,33]. Synergistic interactions have also been observed with flupirtine and the atypical opioids, tramadol and tapentadol, which exhibit the dual mechanisms of action of μ-opioid receptor agonism and inhibition of noradrenaline reuptake[33,34]. Such co-administration will offer benefits such as enhanced analgesic efficacy and prolonged analgesic duration,relative to the minimization of adverse effects and reduction in opioid tolerance.
ANTICONVULSANT
Disruption of the excitatory-inhibitory balance in brain neural networks leading to synchronous activation and recurrent seizures characterises epilepsy. Consistent with its ability to suppress neuronal hyperexcitability flupirtine has demonstrated anticonvulsant activity in the Antiepileptic Drug Development program[7].
Studies have demonstrated flupirtine to be very effective against neonatal seizures induced, for example, by hypoxia/ischaemic injury or chemoconvulsants, with efficacy preferable to current anticonvulsant therapies, such as phenobarbital and diazepam[35-37]. Pretreatment with flupirtine prevented development of neonatal electroclinical seizures, whilst administration after the generation of a seizure prevented subsequent seizures and thereby reduced overall seizure burden. In the immature brain compared to that of an adult the GABAergic inhibitory system is underdeveloped and has fewer GABAAreceptors, different GABAAreceptor subunit composition (e.g., low levels of delta subunits) and lower GABA-mediated currents[38,39]. A decreased efficacy of drugs that target GABAAreceptors such as phenobarbital against neonatal seizures is consistent with the brain underdevelopment[38,39]. As described previously, flupirtine evokes stabilization of neuronal hyperexcitability due to activation of Kv7 and GIRK channel activity, and potentiation of GABA responses of the δ-subunit containing GABAAreceptor[24,27].Although the mechanism responsible for the anti-neonatal seizure activity of flupirtine is unknown, involvement of Kv7 activation is the more likely explanation of providing greater efficacy than and thereby advantage over GABA receptor modulating drugs.
In neonatal rats, flupirtine induced a burst suppression-like electroencephalography (EEG) pattern[36]. Burst suppression is when high voltage activity alternating with periods of no activity in the brain characterize the EEG pattern. Refractory status epilepticus can be terminated by the induction of the burst suppression pattern by midazolam, and as observed with flupirtine[40]. In rat models of established status epilepticus initial data indicated that the combination of flupirtine and diazepam terminated seizures preferentially to either drug alone,however efficacy of flupirtine alone appeared dependent on the animal model[41].Thus, the potential benefit in the clinic available from treatment with flupirtine may be dependent on the underlying aetiology of established status epilepticus.
Febrile seizures are the most common convulsive events in infants and young children where recurrence may be a risk factor for greater likelihood of later epilepsy[42]. Repetitive febrile seizures (RFS) have been associated with impaired hippocampus-dependent long-term memory[43]. Current anti-convulsant drugs,including diazepam, phenobarbital and sodium valproate, although have proven effective at reducing seizure recurrence are limited due to adverse effects[44]. In a rat model of RFS, flupirtine suppressed seizures and reduced risk of further seizures[45].Further, flupirtine was effective against RFS-induced learning and memory impairment and reduced RFS-induced neuronal degeneration. In this RFS model improvement due to flupirtine treatment was greater than that observed with current treatment for recurrent febrile seizures, phenobarbital[45]. Activation of Kv7 channels is the probable property responsible for the efficacy of flupirtine for the treatment of RFS. Consistent with this conclusion is the observation that the expression level of Kv7.2 subunits is low in neonatal neurons and after the first post-natal week increases, which inversely correlates with the incidence of febrile seizures decreasing with age[46].
NEUROPROTECTION
Neuroprotective activity has been exhibited by flupirtine in a variety of neurodegenerative disease models and clinical trials with suggestion of utility as a therapeutic approach in conditions such as Alzheimer’s disease, Parkinson’s disease,Creutzfeldt-Jakob disease, prion disease, age-related macular degeneration and Batten disease[2-4,47,48]. Indirect antagonism of the NMDA receptor and thereby glutamateinduced intracellular Ca2+increase, upregulation of the antiapoptotic protein B-cell lymphoma 2 (Bcl-2) and antioxidant activity via increased glutathione levels and reduced reactive oxygen species levels have all been suggested to be involved in the neuroprotective properties of flupirtine[2-4].
In experimental models of stroke flupirtine evoked neuroprotection when administered before or up to 9 hours post induction of cerebral ischaemia[49-51]. An important signalling pathway contributing to post-ischaemic proteolysis and cell death is an NMDA-induced intracellular calcium increase leading to activation of calpain[52]. Calpain is involved in the degradation of signal-transducer-and-activatorof-transcription-6 (STAT6) which in healthy brains inhibits c-Jun-N-terminal kinases(JNK) and nuclear factor-κB (NF-κB) signalling pathways which are critical to the progression of ischaemic brain injury[51]. As a consequence of indirect NMDA receptor antagonism flupirtine at clinically relevant concentrations reduces the calcium dependent calpain activation and restores the STAT6-induced inhibition of JNK and NF-κB pathways and proteasomal activity[51]. Thus, following flupirtine treatment infarct volumes were reduced, the blood brain barrier integrity stabilized and the inflammatory response and oxidative stress within the ischaemic lesion site reduced.
Chronic stress increases the susceptibility of neurons in the brain to injury with the induction of apoptosis particularly within the hippocampus, which has been proposed to contribute to impaired brain function and stress-related cognitive deficits[53]. In a chronic stress model, flupirtine prevented impairment of spatial learning and memory, alleviated neuronal apoptosis and the reduction of dendritic spine density in the hippocampus[54]. Flupirtine reversed the chronic stress-induced increased expression of the pro-apoptoic regulator Bax, inactivation of the protein kinase B (Akt)/glycogen synthase kinase-3β (GSK-3β) pathway, which regulates learning and memory, synaptic plasticity and cell survival, and reduction in the extracellular signal-regulating kinase 1/2 signalling pathway, which plays a role in cognitive processing[54]. The role of Kv7 channels and/or GABAAreceptors in the effects of flupirtine in chronic stress however need clarification. Interestingly,flupirtine has also been shown to prevent acute stress-induced impairment of spatial memory retrieval and hippocampal long-term potentiation[55]. Activation of Kv7 channels by flupirtine was suggested to be responsible for the reduction in the acute stress-induced impaired memory formation.
Bcl-2 protein complexes with beclin 1 leading to an inhibition of autophagy, thus raised expression of Bcl-2 will influence proteostasis and cell death in neurodegenerative diseases[56]. The Prp106-126 fragment of the prion protein, which is toxic to cortical neurons and involved in the development of prion diseases, reduced levels of the anti-apoptotic proto-oncogene Bcl-2 and of glutathione[57]. Flupirtine blocked the decrease in glutathione and induced the expression of Bcl-2 reducing cell toxicity due to prion protein exposure[57]. Consistent with these findings the cognitive decline in patients with Creutzfeldt-Jakob disease, in a randomized double-blind clinical trial, was decreased following treatment with flupirtine although there were no significant effects on survival[58].
SKELETAL MUSCLE RELAXANT
Flupirtine evokes a reduction in skeletal muscle rigidity and akinesia by the suppression of spinal mono- and polysynaptic reflexes mediated by NMDA receptors[59,60]. The monosynaptic Hoffmann reflex (H-reflex), which does not involve NMDA receptors, was not influenced by flupirtine[59]. These properties of flupirtine are compatible as a therapeutic approach to the treatment of muscle rigidity,spasticity and related musculoskeletal conditions.
The pharmacological properties of flupirtine have also been identified useful in the treatment of myotonia[61,62]. Myotonia and myotonic membrane hyperexcitability induced by anthracene-9-carboxylic acid in murine skeletal muscle was reduced by flupirtine following the activation of Kv7 channels[61]. All KNCQ isoforms are expressed in murine myoblasts with Kv7.2 and Kv7.3 localized at the level of intracellular striations and Kv7.4 subunits restricted to the sarcolemmal membrane[63].Peripheral nerve hyperexcitability associated with neuromyotonia or myokymia is a consequence of inherited mutations in the human KCNA1 gene, which encode juxtaparanodal Kv1.1 channels, or acquired abnormal autoantibodies targeting Kv1 channel subunits[64,65]. A synergy between Kv1 and Kv7 channels in regulating neuronal excitability has been observed and activation of Kv7 channels with flupirtine can reverse the axon hyperexcitability mediated by Kv1.1 channel deficiency[62]. Thus,flupirtine could be a novel therapeutic approach to Kv1-related conditions involving peripheral nerve hyperexcitability.
SMOOTH MUSCLE RELAXATION
Kv7 channels have been identified as important regulators of contractility of vascular,gastrointestinal, urogenital, and tracheobronchial smooth muscle[66]. In smooth muscle cells, Kv7 channels are basally active contributing to stabilizing the resting membrane potential at lower levels and counter depolarizing stimuli, thus reducing cell excitability and contractility. Kv7 channels contribute to the resting membrane potential in smooth muscle cells due to their slow inactivation and low voltage threshold for activation[66].
It is well established that resting membrane potential and contractility in various blood vessels is regulated by Kv7 channels[67]. Expression of KCNQ1, KCNQ4 and KCNQ5 has been reported in a diversity of human, rat and mouse arteries and veins,with KCNQ2 and KCNQ3 expression observed less frequently[66-68]. Many pathological conditions, such as pulmonary hypertension and hyperglycaemia, have been associated with altered expression and function of Kv channels in coronary artery myocytes[69-71]. In addition, reduced expression of Kv7.4 channels have been observed in coronary, renal, and mesenteric arteries of hypertensive rats[72]. Flupirtine evoked concentration-dependent relaxation of rat left and right coronary arteries, with an increase in potassium channel current amplitude and membrane hyperpolarization in myocytes from left coronary arteries but not from right coronary arteries[73]. The differential effects of flupirtine on coronary arteries may be associated with the expression of Kv7.5 being significantly higher in left than in right coronary arteries,whilst Kv7.4 expression was similar. The flupirtine-induced relaxation of porcine left circumflex coronary artery was reduced following endothelial denudation[68]. The flupirtine-induced relaxant response in intact, but not that in artery denuded of endothelium, was inhibited by the non-selective Kv7 channel antagonist linopirdine consistent with the activation of Kv7 channels by flupirtine. In contrast, structurally different Kv7 channels activators (S)-1, retigabine, BMS-254352 and ML213 evoked linopirdine-sensitive relaxations of precontracted porcine and rat coronary arteries that were not affected by removal of the endothelium[74,75]. In porcine left coronary arteries immunohistochemical staining for Kv7.4 was prominent in the intimal(endothelial) layer, but less in the medial (smooth muscle) layer[68]. Strong Kv7.4 immunostaining however was observed in the medial and intimal layers of porcine right coronary artery, and Kv7.5 immunostaining was strong only in the intimal layer with much weaker intensity in the intimal layer[75]. The differential endothelialdependent effects may be due to the distribution and density of channel subtype expression combined with potential subtype selectivity profile of flupirtine relative to other Kv7 channel activators. Thus, the vasorelaxant response to flupirtine in coronary arteries appears to involve endothelium based Kv7 channels and an as yet unidentified mechanism in the smooth muscle cells.
Flupirtine evoked pulmonary vasodilation in lungs from hypoxic rats but not from normoxic rats[76]. Consequently, raised pulmonary vascular resistance due to hypoxia could be prevented and reversed by flupirtine[76,77]. Hypoxia leads to the downregulation of Kv7.4, but not Kv7.1 nor Kv7.5, channels and smooth muscle cell depolarisation. Although the molecular target responsible for the actions of flupirtine have not been identified, activation of the residual Kv7.4 channels, of Kv7.5 channels or a combination of Kv7.4/7.5 needs further evaluation. The Kv7 activation exhibited by flupirtine would be a potential treatment approach for pulmonary hypertension.
Activation of Kv7 channels in bladder smooth muscle cells will dampen electrical activity and regulate excitability. In guinea pig bladder detrusor smooth muscle flupirtine hyperpolarized the cell membrane with a simultaneous cessation of spontaneous transient depolarizing electrical activity and suppression of myogenic spontaneous contractions[78]. Takagi and Hashitani (2016) observed that flupirtine abolished spontaneous action potential discharge in guinea pig detrusor smooth muscle cells without hyperpolarization with the suggestion that only modest opening of Kv7 channels is required[79]. Interestingly Kv7 channels were suggested to only be partial responsible for the flupirtine-induced reduction of the amplitude of spontaneous action potentials associated with spontaneous activity of guinea pig muscularis mucosae[80]. Flupirtine, following Kv7 channel activation, evoked relaxations of the human detrusor muscle, of which overactivity is frequently present in overactive bladder syndrome[81]. As with the guinea pig smooth muscle cells, all five KCNQ genes which encode Kv7.1-7.5 channels are expressed in the human detrusor with the Kv7.4 channel subunit showing the highest expression level[81,82].Thus, selective Kv7.4 channel activators would be preferential novel therapeutic treatments of urinary bladder overactivity conditions. Human overactive bladder or detrusor overactivity in animal models are characterized by increases in spontaneous activity, thus ideal treatment strategy of overactive bladder would be modulation of the spontaneous excitability[83].
All KCNQ isoforms, except KCNQ5, are expressed in human myometrial smooth muscle tissue with a predominance of KCNQ4 and KCNQ1[84]. Thus, the expression profile of KCNQ genes in the uterus resembles other smooth muscles. Although all KCNQ isoforms were expressed in myometrium from pregnant mice in early and late gestation, KCNQ1, KCNQ2, KCNQ4 and KNCQ5 all up-regulated in late pregnancy[83]. Flupirtine suppressed myometrial contractile activity, with a partial effect in early gestation animals and a profound action in late gestation mice[84].Although no teratogenic effects of flupirtine are known or have been reported in animal studies, the potential risk for humans is unknown[85]. Thus, Kv7 channel activation by flupirtine could be a novel approach for tocolysis for use in human preterm labour.
Flupirtine has exhibited relaxing properties of gastrointestinal smooth muscle such as rat stomach and human taenia coli[86,87]. In gastrointestinal tract tissues from animals and humans Kv7.4 and Kv7.5 isoforms had the highest expression levels, with the former being the predominant channel[88]. Thus, this property of flupirtine would be relevant for the treatment of gastrointestinal disorders such as functional dyspepsia and irritable bowel syndrome[88].
Flupirtine has been shown to enhance Kv7 currents in guinea pig airways smooth muscle cells and evoke relaxation of precontracted human airways thereby indicating utility as an effective bronchodilator[89]. In human airways smooth muscle cells expression of KCNQ1, KCNQ4 and KCNQ5 has been reported, whilst KCNQ2 and KCNQ3 were undetectable[90]. In mouse and rat tracheal smooth muscle cells where the expression of KCNQ subtypes reflects that observed in human tissue, flupirtine evoked Kv7 channel dependent hyperpolarization and relaxation[91]. Interestingly,KCNQ2 was the most abundant isoform detected in guinea pig airways smooth muscle cells, suggesting this species may not predict clinical efficacy of flupirtine and similar drugs[89].
AUDITORY AND OCULAR PROPERTIES
Reduction in Kv7.2 and Kv7.3 channel activity leading to hyperactivity in the dorsal cochlear nucleus (an auditory brainstem nucleus) has been associated with the initiation of the auditory disorder tinnitus[92]. Flupirtine inhibited the spontaneous activity in mouse auditory cortical networks which was suggested to involve Kv7 channel activation[93]. These findings are consistent with the pharmacological properties of flupirtine being relevant to the treatment of tinnitus.
Retinal ganglion cell hyperactivity is associated with photoreceptor dystrophic disorders and blindness. Flupirtine decreased spontaneous firing and sensitivity to optogenetic stimulation in mice retinal ganglion cells sensitive to channelrhodopsin2 stimulation[94-96]. Thus, an increase in activity or expression of Kv7 potassium channels,the specific subtypes as yet have not been identified, in retinal amacrine cells would result in decreased spontaneous activity and could be a therapeutic approach to visual disorders.
ADVERSE EFFECTS AND TOLERABILITY
Most common adverse effects associated with flupirtine treatment (i.e., dizziness,drowsiness, nausea, dry mouth, heartburn, fatigue, headache) which occurred within 6 mo of treatment were reported to be mild and transient[2-4]. Consequently flupirtine has been viewed as well tolerated as reflected by regular use since it was first approved in the 1980s.
Long-term flupirtine treatment has been associated with liver injury with rare cases of liver failure and fatality[97,98]. Complex hepatic metabolism of flupirtine involving N-glucuronidation, hydrolysis and N-acetylation leads to the formation of an active metabolite D13223[99,100]. Flupirtine is hydrolysed by carboxylesterase and N-acetyltransferase 2 (NAT2) appears responsible for the acetylation of the hydrolysed metabolite of flupirtine[100]. Reactive quinone diimines, which conjugate with glutathione, however appear to also be formed from the hydrolysed metabolite through non-enzymatic conversion. The quinone diimine intermediates of flupirtine,like the quinone imines intermediates formed with paracetamol and diclofenac, are candidates for the cellular toxicity[100-102]. Thus, acetylation by NAT2 or conjugation by glutathione detoxify by preventing the build-up of quinone diimines[100]. The genetic polymorphisms of NAT2 which show a large inter-individual variability due to genetic polymorphisms and thereby variation in acetylation efficiency could be critical for flupirtine-induced liver injury[100,103]. Humans that exhibit high carboxylesterase activity with slow NAT2 acetylation and have low hepatic glutathione stores appear to be highly susceptible to flupirtine-induced liver injury[100].Other risk factors have been proposed such as single nucleotide polymorphisms of human leukocyte antigen and myeloperoxidase[99,104].
The flupirtine related hepatotoxicity leads to extensive perivenular (zone 3)necrosis with infiltration of ceroid-pigment containing macrophages and lymphocytes[97]. Vulnerability of zone 3 hepatocytes to necrosis appears to be related to stores of glutathione, which detoxifies the reactive quinone diimines, being depleted with a reduced ability to be replenished[97]. N-acetylcysteine, essential for the replenishment of depleted glutathione stores, in combination with prednisolone treatment in patients with flupirtine-induced liver injury has been shown to improve serum biochemistries including alanine aminotransferase and aspartate aminotransferase[105].
The potential of flupirtine-induced liver injury, although rare, led to the European Medicines Agency’s Pharmacovigilance Risk assessment committee issuing risk minimization measures (RMM)[106-108]. As a consequence of limited adherence to the RMM by prescribers flupirtine containing medications were withdrawn from the European market[106].
CONCLUSION
Flupirtine has been used extensively for more than 30 years in the management of pain[2-4]. Effectiveness has been demonstrated in a range of neuronal and non-neuronal biological systems consistent with therapeutic potential as treatment of a diversity of clinical conditions (Table 1). The pharmacological profile exhibited involves actions on several cellular targets, including Kv7 channels, GIRK channels and GABAAreceptors. Although many effects can be accounted for as the result of activation of Kv7 channels, there is suggestion that yet unidentified mechanisms of action may also be involved in the effects of flupirtine.
Flupirtine is providing important information regarding novel mechanistic approaches to the treatment of different clinical conditions. Molecules exhibiting specificity for the pharmacological targets (e.g., Kv7 isoforms) involved in the actions of flupirtine will provide further insight into their clinical potential. Whether the broad-spectrum pharmacology of flupirtine or target-specific actions is preferential to gain benefit, especially in complex clinical conditions, requires further investigation.
Whilst the rare hepatotoxicity associated with flupirtine may limit its use, at risk subjects can be identified by hepatic enzyme screening and risk can be limited by glutathione supplement to replenish stores. Investigation of the oxidative metabolites of flupirtine have been reported to contribute to the activation of Kv7.2/Kv7.3 channels mechanism of action, but not to hepatotoxicity[109]. Thus, the analgesic activity, at least, and cytotoxicity in hepatocytes appear separable in this class of drugs.
In conclusion, flupirtine, in addition to use as an analgesic, shows potential for repurposing as a novel approach to clinical conditions such as tinnitus, visual impairment, memory impairment, requiring neuroprotection or requiring smooth muscle relaxation. Identifying new uses for existing drugs or repurposing bypasses time and cost of drug development. Finally, the pharmacology of flupirtine can also stimulate the identification of next generation related drugs exhibiting target specific actions.
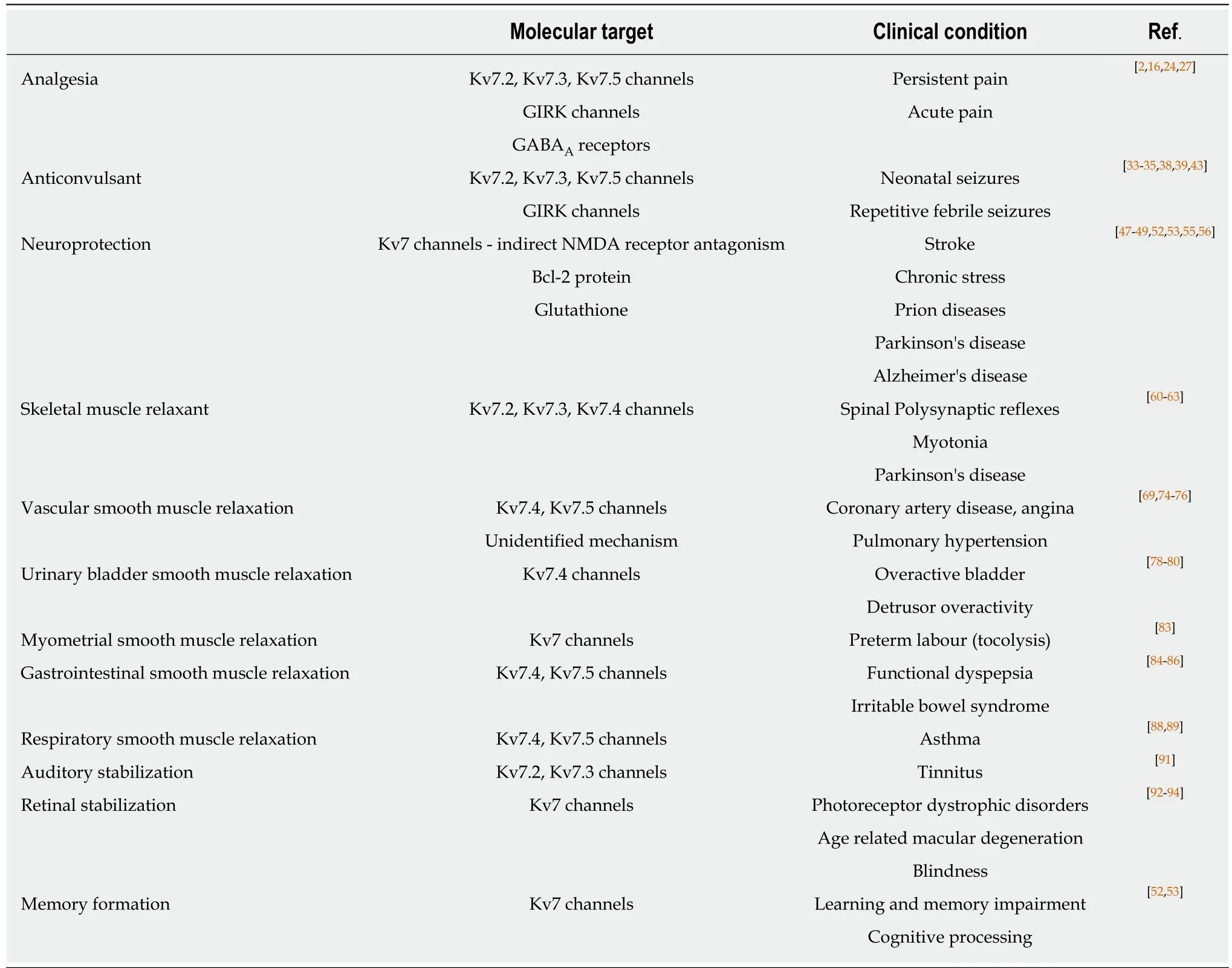
Table 1 Summary of pharmacological profile and potential clinical applications of flupirtine
GIRK: G-protein-regulated inwardly rectifying K+channels; GABA: γ-aminobutyric acid; NMDA: N-methyl-D-aspartate.
