幼年起病、青年期进展至终末期肾病的遗传性肾结石2例
2019-02-12邓海月张琰琴
邓海月 张琰琴 丁 洁 王 芳
研究显示,以往被认为是成人患病的肾结石在儿童中的发生率逐渐上升[1],和成人肾结石的危险因素不同,约1/3~1/2的儿童肾结石患者是由于尿代谢异常所致[2-3],并且与复发性肾结石相关[4]。Daga等[5]的研究显示,25岁前发生肾结石或表现为孤立的肾钙质沉着症的患者中,基因突变者占29.4%。目前已报道的经基因检测诊断为遗传性肾结石的病例多在儿童期以血尿、蛋白尿和(或)双肾结石为首发症状起病,部分患者接受了多次外科碎石治疗,但效果不佳,结石易复发,甚至有患者在儿童期就已进展至终末期肾病(ESRD)[6-9]。为进一步提高临床医师对遗传性肾结石的认识,本文对2例幼儿期起病,青年期进展至ESRD的遗传性肾结石患者的临床及基因资料进行总结和分析。
对象和方法
研究对象2例患者均为幼儿期出现肾结石,青年期进展至ESRD,回顾性分析这2例患者的临床资料。患者外周血基因组DNA均已经目标区域捕获二代测序检测分析和Sanger测序验证。基因检测均征得研究对象或其父母知情同意,并经北京大学第一医院伦理委员会批准。
研究方法留取患者及其父母的外周血2 ml,使用QIAGEN试剂盒(FlexiGene DNA Kit,51206)进行外周血DNA的提取。利用Nimblegen SeqCap EZ Choice系统,针对已知遗传性肾结石/肾钙质沉着相关基因(AGXT、APRT、CA2、CASR、CLCN5、CLDN16、CLDN19、CYP24A1、GRHPR、HNF4A、HOGA1、KCNJ1、OCRL、SLC12A1、SLC26A1、SLC2A9、VDR等)的外显子和侧翼30 bp内含子序列设计靶向测序DNA捕获探针,由天津华大在HiSeq2000或Hiseq2500 (Illumina)平台上进行靶向测序并进行双端短读序。利用BWA(Burrows Wheeler Aligner http://sourceforge.net/projects/biobwa/)将过滤后数据对比人类基因组(hg19)。使用SOAPsnp (http://soap.genomics.org.cn/)和SAM(http://sourceforge.net/projects/samtools/ )分别检测单核苷酸变异和小片段插入缺失,拷贝数变异通过华大batCNV1.0识别。
去除支持读数小于10,比率<10%的变异,在对照数据库如NCBI dbSNP build141(http://www.ncbi.nlm.nih.gov/SNP/),1000 Genomes Project(http://www.1000genomes.org/),Exome sequencing project (ESP6500.http://evs.gs.washington.edu/EVS/),Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/)和华大内部数据库中发现的最小等位基因频率≥1%的变异作为多态被过滤掉。使用Mutation Taster、SIFT、PolyPhen 2和Condel预测未报道变异的功能意义,应用Human Splicing Finder 3.0预测变异位点的剪切功能。利用PhyloP Primates工具评估变异位点的进化保守性,根据The American College of Medical Genetics and Genomics (ACMG)遗传变异分类标准指南对变异进行评价。
临床资料
例1 男,汉族,1岁时因排尿困难就诊发现双肾多发结石,2岁因结石梗阻尿道行膀胱切开取石术,6岁时先后行2次体外冲击波碎石,自行口服中药至9岁,后未行特殊处理,未定期复查。20岁时因右侧腰腹部疼痛10余小时就诊于当地医院。查体右肾区叩痛,右侧输尿管行径区中下段深压痛。实验室检查:(1)血常规示白细胞6.05×109/L,血红蛋白(Hb)101 g/L;(2)血生化示钾4.2 mmol/L,镁0.56 mmol/L,钙1.76 mmol/L,磷2.26 mmol/L,血清肌酐(SCr)1 238 μmol/L,尿素氮(BUN)56.9 mmol/L,尿酸(UA)640 μmol/L,胱抑素C 5.1 mg/L;(3)尿常规示蛋白+,未进行尿沉渣镜检;(4)24h尿蛋白定量为1.9 g/24h;(5)尿液结晶分析及中段尿培养阴性;(6)泌尿系CT示双肾外形变小,肾实质变薄,双肾多发结石,右输尿管下段结石并右肾及输尿管积水。于当地医院行右输尿管镜下激光碎石及血液透析(HD)治疗。
患者父母非近亲结婚,家庭成员中均无相关肾结石病史。因考虑其肾结石病因为家族性低镁血症伴高钙尿症和肾钙质沉着症(FHHNC)可能性大,经患者知情同意后,分别提取患者及父母外周血DNA,采用目标序列靶向捕获测序技术进行遗传性肾结石相关的基因分析,异常结果均经Sanger测序验证。患者检出CLDN16(NM_006580.3) Exon 4:c.715G>T(p.Gly239*)纯合变异,父母分别检测到相同杂合变异(图1)。该变异未在HGMD和ClinVar数据库中检索到,为首次报道。在正常人群数据库《1 000 Genomes Project》、《ExAC》和《ESP》中无记录。第239位甘氨酸在脊椎动物中高度保守,Mutation Taster预测变异有害。根据ACMG遗传变异分类标准指南,c.715G>T (p.Gly239*)变异为致病的,依据患者病史及基因检测结果诊断为FHHNC。

图1 例1及其父母CLDN16基因测序结果
例2 女,汉族,1岁时出现尿中排黄白色结石,家长未予干预。7~8岁时多次X线片提示双肾结石(图2),曾行结石成分分析提示“草酸钙结石”。22岁时因“纳差、恶心3个月”就诊于当地医院,血常规示Hb 85 g/L,血生化示SCr 1 262.4 μmol/L,BUN 36.8 mmol/L,泌尿系超声示双肾小伴弥漫多发结石,予HD治疗。获得基因检测结果后行肝肾移植。
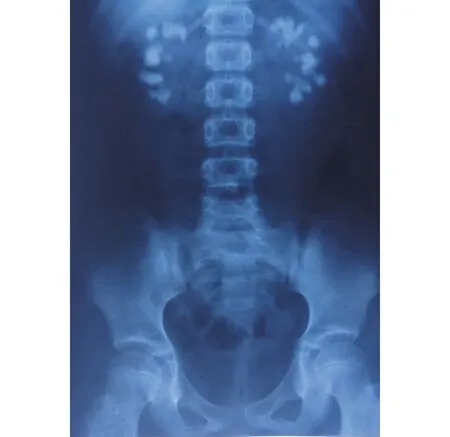
图2 例2腹部X线片(8岁)示双肾结石
患者母亲、外祖父和祖父均有肾结石病史,肾功能均正常。其中患者母亲在35岁时发现肾结石,自行排出后无复发。患者外祖父、祖父分别在64岁、60岁发现肾结石,手术碎石后无复发。其父否认肾结石病史。因考虑该患者原发性高草酸尿症(PH)可能性大,经其知情同意后,分别提取患者及父母外周血DNA,采用目标序列靶向捕获测序技术进行遗传性肾结石相关的基因分析,异常结果均经Sanger测序验证。患者检出AGXT(NM_000030.2)复合杂合变异:Exon 1:c.32C>G(p.Pro11Arg,父源)和Exon 2:c.346G>A(p.Gly116Arg,母源)(图3)。这两个变异均已被报道[10-11],在正常人群数据库1000 Genomes Project和ESP中的频率均为0,ExAC中频率分别为0.000 157 6和0,Mutation Taster、SIFT和PolyPhen预测均为有害,而且第11位Pro和116位Gly在脊椎动物中均高度保守。根据ACMG遗传变异分类标准指南,评价c.32C>G(p.Pro11Arg)和c.346G>A(p.Gly116Arg)变异为致病的和可能致病的。考虑患者病史和该基因变异高度相关,因此诊断为PH 1型。
讨 论

图3 例2及其父母AGXT基因测序结果A:例2及其父亲检测到AGXT基因 Exon 1:c.32C>G(p.Pro11Arg)突变;B:例2及其母亲检测到AGXT基因Exon 2:c.346G>A(p.Gly116Arg)突变
虽然儿童肾结石的发病率低于成人,但是代谢异常引起的肾结石在儿童中比成人更常见,且可导致肾结石复发,进而给患病家庭带来沉重的经济负担。近年研究显示[5],至少30个基因突变可致肾结石,遗传方式包括常染色体隐性遗传、常染色体显性遗传和X连锁遗传。16.8%~20.8%的儿童期起病的肾结石或肾钙质沉着症以及11.4%的成年期起病的肾结石或肾钙质沉着症为单基因突变所致[12-13]。然而由于临床医师对遗传性肾结石的认识不足,往往导致对此类疾病的诊治存在延误。如本研究中2例患者所示,均在1岁以肾结石起病,且结石反复发作,直至20岁左右进展至ESRD时经基因检测才明确诊断为单基因突变导致的遗传性肾结石,已丧失了延缓肾功能进展的治疗机会。特别是例2,结石分析显示草酸钙结石时便应高度怀疑PH,应明确诊断并予以干预,遗憾的是发展至ESRD时方考虑寻找肾结石病因。因此,对于儿童期起病的肾结石患者,后天因素(尿路感染、药物或疾病相关)不能解释时应注意寻找遗传因素。
低镁血症、高钙尿症和肾钙质沉着症是诊断FHHNC的三联征[14]。该病呈常染色体隐性遗传。例1患者1岁起病,20岁ESRD时表现为低镁血症及双肾和输尿管结石,因此在缺少24h尿钙检测结果的情况下临床高度怀疑FHHNC,最终经基因检测明确诊断为CLDN16基因突变所致FHHNC(OMIM# 248250)。由于CLDN19基因突变所致FHHNC的患者通常具有眼部受累的表现,如近视,眼球震颤和黄斑缺损[15-16]。因此对怀疑FHHNC的患者除了要注意肾脏改变外,也需完善眼科检查,从而为明确诊断提供线索。
FHHNC患者肾功能的减退可能受种族、致病基因等多种因素影响。Weber等[17]的研究显示FHHNC患者终末期肾病的中位年龄为14.5岁。Kari等[18]的研究显示阿拉伯裔FHHNC患者较欧洲裔患者进展至慢性肾脏病的速度缓慢,Godron等[14]的研究显示CLDN16基因突变患者较CLDN19基因突变的患者肾功能恶化慢。FHHNC的治疗目的是延缓肾功能进展及对肾结石的常规管理,目前尚无有效的药物治疗措施。补充大剂量的镁制剂和噻嗪类利尿剂往往被用于减少尿钙排泄和肾脏钙质沉着,但是一些研究显示[14,17],噻嗪类药物并未能延缓肾功能的进展。ESRD的FHHNC患者最佳治疗措施是肾移植,移植肾可纠正其肾小管对镁和钙离子转运的缺陷,使FHHNC不复发[16,19]。
儿童或青少年出现双肾草酸钙结石时应考虑PH的可能[20]。该病呈常染色体隐性遗传,分为3型,致病基因分别为AGXT、GRHPR和HOGA1。其中以PH 1型(OMIM# 604285)最常见,占80%[21]。除肾结石和慢性肾脏病外,PH 1型患者可出现心脏、骨、关节、视网膜、皮肤等部位的草酸钙沉着[22]。不同于FHHNC,PH 1型存在有效的内科治疗措施:作为唯一被证明能够减少草酸盐排泄的药物,维生素B6对约50%的PH 1型患者有效,特别是具有p.Gly170Arg和p.Phe152Ile突变者[23-24]。因此,PH 1型患者获得早期诊断和早期干预可以延缓肾功能的进展。此外,摄入足够量的液体[2~3 L/(m2·24h)]、限制饮食中草酸盐的摄入以及口服柠檬酸盐或中性磷酸盐可减少PH草酸钙结晶形成[20]。例2患者1岁发病,结石成分分析示草酸钙结石,基因检测示AGXT基因复合杂合突变,故诊断PH 1型明确。遗憾的是该患者未得到及时诊治,致22岁进展到ESRD。此外,虽然该患者家系中多人患肾结石,但其结局和起病年龄与例2患者均不同,并且例2母亲仅检测到AGXT基因的一个杂合突变,考虑家人肾结石形成原因系后天因素可能性大[25],提示临床实践中一个家系多人患肾结石时需分析是否是同一原因所致。
已知AGXT基因突变致肝细胞丙氨酸-乙醛酸盐转氨酶活性下降或缺乏,进而引起草酸合成增加、在体内多器官沉积,因此PH 1型患者进展至ESRD时单纯肾脏移植后移植肾仍会发生草酸钙沉积,而肝肾联合移植则避免了这一问题。可见明确PH诊断尚有助于指导器官移植。
总之,对于儿童期起病的肾结石患者,需要警惕遗传因素,通过基因检测可协助尽早明确诊断并指导治疗。
