Deciphering Brain Complexity Using Single-cell Sequencing
2019-02-08QuanhuaMuYiyunChenJiguangWang
Quanhua MuYiyun ChenJiguang Wang*c
Department of Chemical and Biological Engineering,Division of Life Science,Center for Systems Biology and Human Health and State Key Laboratory of Molecular Neuroscience,The Hong Kong University of Science and Technology,Clear Water Bay,Kowloon,Hong Kong Special Administrative Region,China
Abstract The human brain contains billions of highly differentiated and interconnected cells that form intricate neural networks and collectively control the physical activities and high-level cognitive functions,such as memory,decision-making,and social behavior.Big data is required to decipher the complexity of cell types,as well as connectivity and functions of the brain.The newly developed single-cell sequencing technology,which provides a comprehensive landscape of brain cell type diversity by profiling the transcriptome,genome,and/or epigenome of individual cells,has contributed substantially to revealing the complexity and dynamics of the brain and providing new insights into brain development and brain-related disorders.In this review,we first introduce the progresses in both experimental and computational methods of single-cell sequencing technology.Applications of single-cell sequencing-based technologies in brain research,including cell type classification,brain development,and brain disease mechanisms,are then elucidated by representative studies.Lastly,we provided our perspectives into the challenges and future developments in thefield of single-cell sequencing.In summary,this mini review aims to provide an overview of how big data generated from single-cell sequencing have empowered the advancements in neuroscience and shed light on the complex problems in understanding brain functions and diseases.
KEYWORDS Neuroscience;Single-cell RNA-seq;Cell type;Brain development;Brain diseases
Introduction
The complex cellular diversity and connectivity within brain cells are fundamental to the function of human brain.The classification of cell types in the nervous system is first brought into focus by Ramo´n y Cajal’s work published over a century ago[1],which covers only the gross morphology and major classes of neurons and glia but lacks detailed description.In current neuroscience,combinations of parameters are applied to identify neuronal cell types,which include cell morphology,anatomical location,electrophysiological activities,synaptic properties,connectivity in neural circuits,and expression of certain marker genes.However,the construction of a comprehensive brain cell type atlas,with incorporation of their molecular identity,lineage in development,and contribution to brain diseases,remains a great challenge in the field of brain research.
The development of single-cell technologies,especially single-cell RNA-sequencing(scRNA-seq),has provided new opportunity to address this challenge by looking through transcriptomic profile of each individual cell.Since the first introduction of scRNA-seq technique by Tang et al.in 2009[2],this technology has developed extensively and applied broadly to different biological systems.In recent years,a dozen of scRNA-seq studies that look into the cellular composition,heterogeneity,and disease-specific populations in mammalian brain has also demonstrated the power of this technology in addressing the challenges in understanding the complexity,connectivity,and functions of brain cell types[3,4].As a result,the American Brain Initiative,the European Human Brain Project and also the Chinese Brain Project give top priority to the cell type classification in their endeavors[5-7].In the recently launched Human Cell Atlas project,scientists,are aiming to ‘create comprehensive reference maps of all human cells”using scRNA-seq[8].Apart from elucidating the cell types in brain using scRNA-seq,advancements in other single-cell sequencing technologies,including single-cell genomics,epigenomics(including methylation,DNA accessibility,and chromosome conformation),and multi-omics,have also provided new tools to study the whole brain at single-cell resolution and brought new insights into the developmental lineage,epigenetic markers,and functional states of individual cells[9-13].Moreover,by collecting cells from different spatial locations,temporal points,and disease states,single-cell sequencing has empowered our understanding of the brain development,function,and diseases at an unprecedented depth and resolution.
In this review,we started by summarizing the experimental(see the ‘Advances in single-cell sequencing platforms”section)and computational techniques(see the ‘Advances in computational analysis methods of scRNA-seq data”section)in scRNA-seq,which have boosted its throughput and analytic power.Next,we described the landmark papers as well as recent progress in single-cell sequencing technologies in resolving brain complexity(see the ‘Applications of single-cell sequencing in brain studies”section)in terms of:(1)the diversity and heterogeneity of cell types in the brain,(2)the dynamic changes in brain cell types,expression profi les,and the accumulation of somatic mutations during development and aging,(3)the associations between brain cell types and neuronal diseases,and(4)the contributions of glioma stem cells and macrophages to the intratumoral heterogeneity of brain cancer.Lastly,we provided our insights into the future trends and developments in the field of single-cell sequencing.
Advances in single-cell sequencing platforms
Typical next-generation sequencers require the input DNA to be at a nanogram level,which is orders of magnitude higher than the amount of RNA in one single cell.Therefore,the first challenge in scRNA-seq experiments is the amplification step in sequencing library preparation.In the first paper that introduced scRNA-seq technology in 2009,Tang et al.used a pair of poly(T)primers with anchor sequences to capture the mRNA from a mouse blastomere,and then amplified the reversely-transcribed double-stranded cDNA using two anchor sequences as primers[2].This protocol has stable and elegant performance,and more importantly it inspired innovations of new technologies to expand its applications,such as single-cell universal poly(A)-independent RNA sequencing(SUPeR-seq),quantitative single-cell RNA-seq(Quartz-seq)and single-celltagged reverse transcription sequencing(STRT-seq)[14-16].Smart-seq,which utilizes Moloney murine leukemia virus reverse transcriptase that adds 2-5 untemplated nucleotides to the 3′end of the first cDNA strand,allows the template switch from the first synthesized cDNA strand to the second strand with a helper oligo called template-switching oligo,thus enabling the capture of fulllength transcript[17].Further improvement in sensitivity,accuracy,and full-length coverage in Smart-seq2 makes it a widely-used scRNA-seq library preparation protocol[18].Apart from PCR-based amplification methods mentioned above,other methods have been established for amplification by in vitro transcription[19-21],and are applied to various platforms[22-24].
Apart from the single-cell transcriptome library preparation protocols,the revolution in automatic cell separation platforms has also enabled the exponential scale-up in the number of single cells sequenced in recent years,which can go up to hundreds of thousands of single cells per study[25].Moving from manual selection and pipetting[2],several automated single-cell compartmentalization methods have been developed.Methods that isolate single cells into separated wells using fluorescence-activated cell sorting(FACS)or robotic arms have speeded up the single cell isolation[21,26].Microfluidic platforms,such as the Fluidigm C1 system,isolate single cells on a chip,where single cells are passively captured into 96 isolated chambers[27].While the method also overcomes the laborious reagent adding steps,the total number of cells captured by the single-use microfluidic chip limits the throughput of this method.Alternative methods that randomly capture single cells with barcoded beads using microfluidic droplet generators,such as Droplet sequencing(Drop-seq)[28],indexing droplets RNA sequencing(inDrop)[23],and GemCode/Chromium 10× (widely known as 10× Genomics)[29]stand out by their high throughput and low cost.Nonetheless,these methods have limited sequencing depth and can only reveal the 3’end sequence of transcripts.Picoliter wells that capture single cell with barcoded beads have also been developed[24,30,31],with recent improvements in Microwell-seq that further reduce the cost and rate of capturing cell doublets[32]. Moreover, split-pool ligation-based transcriptome sequencing(SPLiT-seq)has been recently developed and,by multiple rounds of split-pool barcoding,the cost of sequencing per cell is further reduced[33]to an estimated cost of 50 cents/-cell.A similar method called single-cell combinatorial indexing RNA sequencing(sci-RNA-seq)also utilized combinatorial barcoding strategy for single cell demultiplexing[34],and has been optimized to profile over 2,000,000 single cells in a single experiment[35].Apart from the platforms designed to capture individual cells,single-nucleus isolation and sequencing methods,such as single-nucleus RNA sequencing(sNuc-seq)[36]and sNuc-seq with droplet technology(DroNc-seq)[22],generate highly concordant expression data as scRNA-seq while overcoming the requirement for intact cells and the problems of losing neuronal cell types differentially due to cell size heterogeneity.Applied to frozen samples in human tissue banks,single-nucleus RNA sequencing methods have shown to be more promising than the whole-cell RNA-seq[37].Chemical fixation methods may also facilitate stabilization and preservation ofdissociated cellsforweeksbefore scRNA-seq,while producing comparable results as data generated from fresh samples[38].
Recently,several scRNA-seq technologies have been developed to study the structural and dynamic properties of RNA transcripts at single-cell level,or to simultaneously profile multi-omic data in the same cell.For instance,single-cell isoform RNA-seq(ScISOr-Seq)was developed to identify RNA isoforms and splicing sites[39].Droplet-assisted RNA targeting by single-cell sequencing(DART-seq)combined multiplexed amplicon sequencing and transcriptome profiling in single cells,enabling simultaneous determination of virus genotypes and gene expression of the infected cell[40].Combination of fluorescence in situ hybridization with scRNA-seq revealed the connection of spatially associated cells[41].To overcome the limitation that current scRNA-seq provides only a snapshot of the transcription,single-cell,thiol-(SH)-linked alkylation of RNA for metabolic labeling sequencing sequencing(scSLAM-seq)uncovered dynamics of transcriptional activity directly by differentiating between new and old RNA[42].Finally,single-cell triple omics sequencing(scTrio-seq)technique is able to provide information of the mutations,transcriptome,and methylome of single cells[43].Other single cell sequencing platforms for unimodal profiling of the genomic,epigenomic,and chromosome conformation,as well as multimodal measurements of RNA and other components,have been summarized in a recent review by Stuart and Satija[44].
These technological advancements enable automatic,highthroughput single-cell capture,and sequencing,which not only provide new tools for brain research and huge amount of data for analysis,but also inspire and empower future research in generating a comprehensive human brain cell atlas.To provide a practical guide for future research,we summarized the characteristics of common scRNA-seq library preparation methods,by comparing the throughput,transcript coverage,ability of detecting RNA without poly(A)tail,and sensitivity in detecting low abundance genes(Table 1).Several comprehensive reviews have compared the performance of different scRNA-seq platforms.Although these platforms demonstrate great accuracy in transcript level quantifications,their sensitivity for detecting genes with low expression varies[45,46].Additionally,these protocolsgenerate eithercDNA library composed of only the 3′-end for quantification,or full-length transcripts by tagmentation that allow detection of different transcript variants and splicing events among cell types[47,48].Thus,requirements for sensitivity,full-length transcript information,number of cells,and reaction volumes are critical factors for selecting single-cell sequencing platforms to address specific research questions.
Advances in computational analysis methods of scRNA-seq data
A typical workflow of scRNA-seq data analysis consists of preprocessing,data normalization,dimensionality reduction,clustering,differential gene expression,and gene expression dynamics analysis(Figure 1).Although data obtained from scRNA-seq are often structurally identical to the data obtained from bulk RNA-seq,scRNA-seq data have two important features that require special design in the computational methods to distinguish technical noises from true variation signals.These include(1)dropout events that introduce abundant zero values in the gene expression matrix;and(2)high variations in gene expression between cells and/or batches of experiments(also called ‘batch effects’).
The data analysis starts with raw sequencing reads.In the preprocessing step,a process called demultiplexing is performed to assign reads to each cell based on the cell-specificbarcodes.In the presence of unique molecular identifiers(UMIs,short random sequences attached to individual cDNA molecules)at the 5′or 3′end of reads,deduplication of reads is also performed to remove PCR-generated duplicated reads.Then,the barcodes,UMIs,and adaptor sequences are trimmed from the reads,and the clean reads are subsequently mapped to the reference genome.For droplet-based technologies,some droplets may contain two or even more cells,and these ‘doublets’can be computationally identified by demuxlet[49].Quality control should be conducted along all these steps,including removing reads with low quality values,reads that are poorly mapped,and cells that have few high-quality reads.Although some popular tools such as FastQC[50]are widely used,home-brew scripts may also be utilized for preprocessing,depending on the design of the experiments.The final output of preprocessing is the expression values of each gene in the qualified cells,which are represented as read counts.

Table 1 Comparison of scRNA-seq platforms

Figure 1 A typical workflow of scRNA-seq data analysis
A critical step following data preprocessing is normalization,which intends to remove the artificial gene expression variation.Such variation may originate from many sources,including amplification biases,sequencing depth,GC content,capture and reverse transcription efficiencies.Normalization has been demonstrated to greatly affect the downstream analysis such as differential gene expression.For bulk RNA-seq data,global scaling(dividing the read counts by a global scaling factor)is applied to enable comparison between samples.To minimize the effects of the dropout events,similar methods have been developed for scRNA-seq data,where the global scaling factor is adjusted by quantile normalization or using only genes with relatively constant expression across cells.However,the underlying assumption of these methods is that the total RNA amount is identical across all samples and the variation in read counts is solely attributed to sequencing depth,which may not be true for single cells.Additionally,such approaches are often highly unstable,since they can be affected by the abundant zero values in scRNA-seq data.For scRNA-seq experiments,internal control such as synthetic spike-ins(external transcripts added at known concentrations)or UMIs are better options since they can reflect the differences in RNA content and amplification efficiencies between cells.Current scRNA-seq data processing packages,such as Seurat[51]and single-cell analysis toolkit for gene expression data in R(Scater)[52],have internal functions to handle spike-ins and UMIs.Imputation methods,such as Markov affinity-based graph imputation of cells(MAGIC)[53],single-cell analysis via expression recovery(SAVER)[54],and scImpute[55],also demonstrate effective correction of dropout events,as well as recovery of transcript levels and gene-gene associations.Although some biases can be reduced after normalization,other technical and biological variations(such as fluctuations due to different stages in cell cycle)and the batch effects still exist in the data.Several methods are available to deal with batch effects,such as ComBat[56]and mutual nearest neighbors(MNN)[57].When batch information is available,ComBat applies an empirical Bayesian framework to correct the batch effects.MNN first detects mutual nearest neighbors and then adjust the batch effects based on the deviation of the shared subpopulations in each batch.According to the comparison by Haghverdi et al.[57],MNN shows superior performance than ComBat.More recent batch-correction tools include Scanorama[58]and Harmony[59].In practice,careful experimental design that can remove or balance batch effects would be extremely helpful.
After normalization,dimensionality reduction methods are applied to project the high-dimensional(dimensionality as the number of detected genes)measurements of each data point(one data point as one cell)into a low-dimensional subspace to visualize the population composition and discover new subpopulations.The genes are usually filtered by the dispersion of their expression and a few hundreds of most variable genes are selected to capture important features across the population.Principal component analysis(PCA)is efficient and easy to implement,and it is widely used,since the results are highly interpretable.Another linear method is zero-inflated factor analysis(ZIFA)[60],which in essence is a factor analysis method but takes into account the presence of dropouts.To better represent the dropouts,Risso et al.developed a general and flexible model named zero-inflated negative binomial model(ZINB-WaVE)[61].This model inspired the development of two autoencoder frameworks,single-cell variational inference(scVI)[62]and deep count autoencoder network(DCA)[63],fordimensionality reduction oflarge-scale scRNA-seq data.Linear methods assume linear relationship between data variables,but this might not hold true for the gene expression data.t-distributed stochastic neighbor embedding(t-SNE)[64]is a non-linear method,which is optimized to map high dimensional data points into two or three dimensional space,primarily for visualization.Although hard to interpret,the decent results generated by t-SNE make it the current state-of-the-art method to visualize scRNA-seq data.Recently,a method named uniform manifold approximation and projection(UMAP)[65,66],which is based on theories in Riemannian geometry and algebraic topology,has been developed,and soon demonstrated arguably better performance than t-SNE due to its higher efficiency and better preservation of continuum.Another method,single-cell interpretation via multi-kernel learning(SIMLR)[67],applies a multi-kernel learning algorithm to learn a distance metric that better fits the structure of the data.Embedding with t-SNE based on the learned distance metric,Wang et al.have demonstrated good performance of SIMILR on multiple scRNA-seq datasets[67].
Aided by dimensionality reduction,identification of subpopulations of cell types can be achieved by clustering methods.For unsupervised clustering, although traditional clustering methods such as hierarchical clustering and K-means clustering might be used,they are often hindered by the scale and the noise in the data.Clustering through imputation and dimensionality reduction(CIDR)[68]attenuates the effects of dropouts by imputing the zero values before clustering.Recently,a group of graph-based clustering methods,including shared nearest neighbor(SNN)-Cliq[69],rare cell type identification(RaceID)[70],RaceID2[71],PhenoGraph[72],and Seurat[51],has been developed and proved to highly efficient and robust.These methods embed the cells into a graph,with each edge representing the similarity(such as Euclidean distance or Pearson correlation)between the two cells,and then partition the graph into highly interconnected modules.Consensus clustering has also been adopted for scRNA-seq data clustering,and shown to be highly accurate and robust[73].Due to the heavy time consuming nature of consensus clustering,a rule of thumb for unsupervised single cell clustering is to use single-cell consensus clustering(SC3,integrated in Scater[52])when the number of cells is<5000 but use Seurat instead when there are more than 5000 cells.For most cases,however,we have some prior knowledge of the cells(e.g.,major cell types,cell surface markers),and Monocle provides an option to instruct clustering by specifying known cell type markers[47].Although both unsupervised and semi-supervised clustering methods are provided,Monocle recommends the semi-supervised method for more reliable results[74].UNCURL also supervises the clustering by prior biological knowledge[75].BackSPIN,a divisive biclustering method based on sorting points into neighborhoods(SPIN)[76]can cluster genes and cells simultaneously,enabling us to obtain the information on the cell types and meanwhile their gene markers as well.Several computational tools that automatically assign each single cell were available,such as SingleR[77],scScope[78],and CellAssign[79],but all of them rely on cell-type specific markers either from reference databases or input by the user.
Discovering differentially expressed genes has important implications in defining cell types and identifying markers of each subpopulation.However,direct application of traditional methods,such as DESeq[80],might be problematic,because of the presence of abundant zeros in scRNA-seq data.To accommodate the multi-modality in the distribution of gene expression,mixture-model-based approaches,such as modelbased analysis of single cell transcriptomics(MAST)[81],single-cell differential expression(scDE)[82],and single-cell differential distributions(scDD)[83],have been developed,claiming highly improved performance than traditional differential gene expression tools.In a recent study,Soneson and Robinson have compared the performance of scRNA-seq differential expression methods in a consistently processed scRNA-seq data collection named consistent quantification of external RNA-seq data(conquer).They find that traditional methods such as edgeR and voom-limma perform equally well as scRNA-seq-specific methods,if lowly-expressed genes arefiltered out[84].With proper prefiltering,even simple t-testfinds the right differentially expressed genes with low false discovery rate.
Finally,in order to infer the dynamic path of cellular development and/or differentiation from a snapshot of gene expression pattern of individual cells,several pseudotemporal ordering algorithms have been designed.The very first yet efficient and robust method is Monocle[74].In Monocle,the data are first dimension reduced by independent component analysis,then a graph is constructed by adding connecting edges between highly similar cells.The graph is deduced to a maximum spanning tree(MST),and the longest path in the tree is regarded as the evolution path.Branching is opened if alternative trajectories are found when examining cells not along the longest path.Another type of methods is based on theories and algorithms in topological data analysis such as diffusion map and mapper.Single-cell topological RNA-seq analysis(scTDA)[85],for example,starts with dimensionalityreduction by PCA,then splitsthe twodimensional projection into tiles,and builds a tree using the tiles as nodes.The root node is either given or inferred from the tree.Several other methods are developed,including Waterfall[86],Wishbone[87],selective locally linear inference of cellular expression relationships(SLICER)[88],Destiny[89],and Sincell[90].There is complementarity between different methods as detailed by a large-scale comparison of trajectory inference methods[91].Therefore,selecting the proper method should largely rely on knowledge about the dataset.
Applications of single-cell sequencing in brain studies
From the year 2015 onwards,over 80 papers have reported detailed characterization of brain cell types in different brain regions,and at developmental stages or disease status using scRNA-seq(Figure 2 and Table 2)[3,4].In addition to the increasing number of publications,we have also observed an exponentially increasing number of sequenced cells per study in the last 5 years.The technology is not only inspiring more studies in recent years,but also exponentially scaling up the number of single cells profiled in each study,which has empowered the construction of a comprehensive landscape of the cell types in the brain.
Revealing the diversity of brain cell types

Figure 2 The exponential increase of the number of cells sequenced in published scRNA-seq studies of the brain
Large-scale single-cell transcriptome-based classification studies of the nervous system were first conducted in mouse models.Sequencing over 3000 single cells in mouse somatosensory cortex and hippocampus CA1,in one of the landmark papers of scRNA-seq,Zeisel et al.identified nine major brain cell types that can be further grouped into 49 subpopulations.This study has extensively expanded the classical understanding of brain cell taxonomy[92].The early studies are supportive of the hypothesis that,based on single-cell transcriptome characteristics,brain cells can be unbiasedly clustered into similar cell types,presenting a map of cell type complexity and diversity in the brain[93].Droplet-based isolation has enabled high-throughput,unbiased profiling of cell types in mouse nervous systems by scRNA-seq or snRNA-seq[22,94].For instance,more than 500,000 single cells were sequenced by Zeisel et al.[95]and Saunders et al.[96],and more than 1,300,000 cells were sequenced by 10×Genomics(https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.3.0/1M_neurons).These studies provide valuable resources for discovering cell type diversity in mouse brain and peripheral nervous system.Characterization of brain cell types in humans has also provided rich resources for elucidating the transcriptional subtypesand novelmarkergenesin normalbrain [97],assessing in vitro culture models[98,99],and analyzing the evolutionary conservation of cell types by comparing with scRNA-seq data from other species[100-102].

Table 2 Summary of studies that characterize the single-cell transcriptome in the brain

Table 2 Summary of studies that characterize the single-cell transcriptome in the brain

Table 2 (continued)
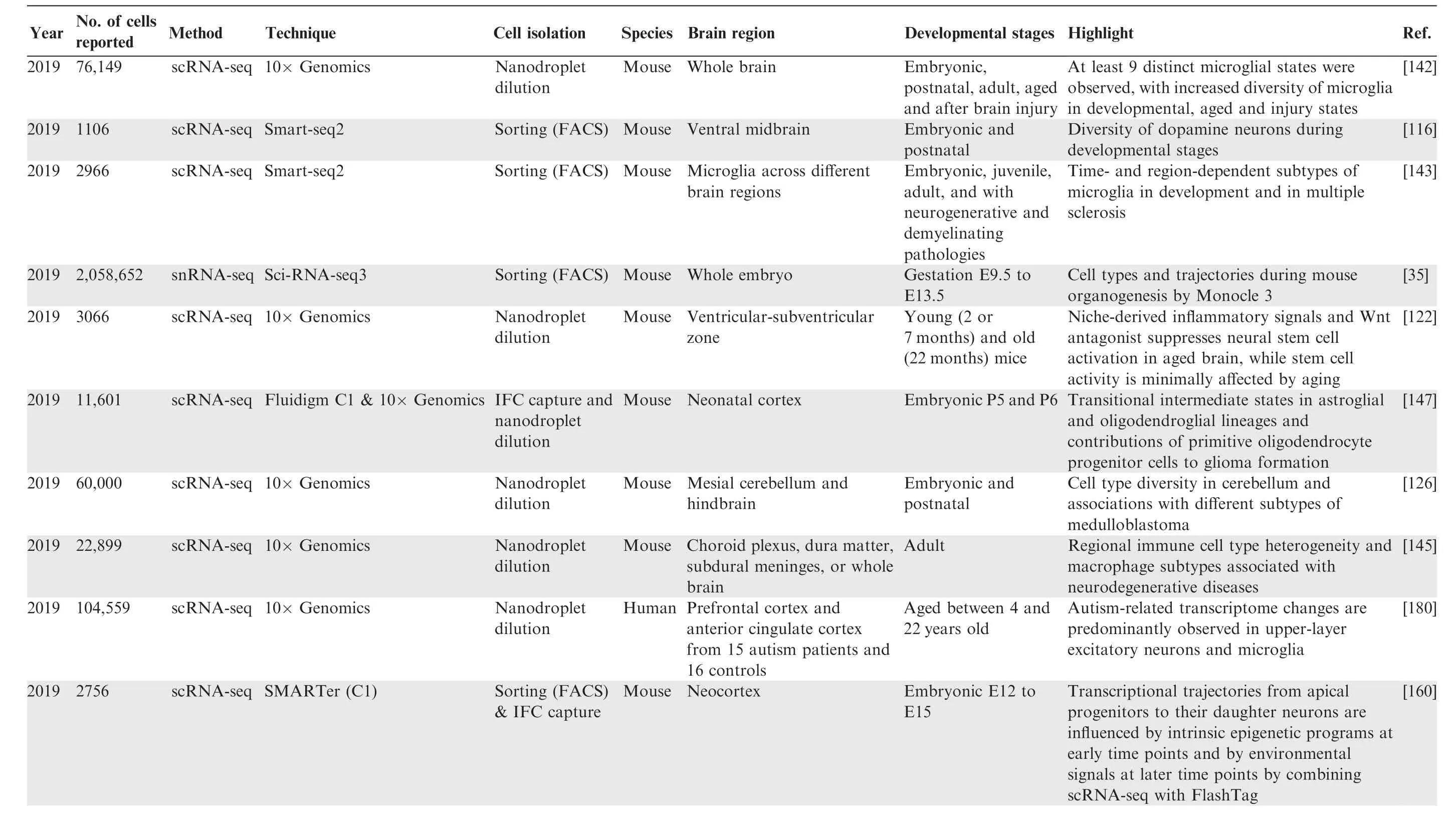
Table 2 Summary of studies that characterize the single-cell transcriptome in the brain

Table 2 (continued)
Brain functions are known to be partitioned into different brain regions,where locally and distally connected neurons coordinate to integrate signals and perform specific tasks.scRNA-seq technology has greatly facilitated research efforts in resolving regional cell type landscapes,including the visual cortex [103-107],motorcortex [107,108],hypothalamus[109-111],amygdala[112],dentate gyrus[113],ganglionic eminence[114,115],ventral midbrain[100,116],preoptic region[117],optic lobe[118],hippocampus[22,36,92,101],subventricular zone and ventricular-subventricular zone[119-124],neural crest[125],and cerebellum[126,127].Moreover,a few studies following a unified set of protocols have been reported to dissect and sequence single cells across multiple brain regions at fetal or adult stages in mice[95,96,128]and in humans[37,129].These studies have enabled comprehensive capture of brain cell types,comparison of regional differences in cell type compositions and expression profiles,as well as mining associations between brain cell types and neurological disorders[130].However,challenges remain to resolve the positional information of individual cells in threedimensional space,as such information is lost when cells are dissociated from intact tissues into single cell suspensions.While several RNA-FISH-based techniques in spatial transcriptomics(reviewed by Crosetto et al.[131]and Lein et al.[132])have been developed and applied to visualize spatial expression patterns of up to 10,000 genes in mouse hippocampus[133],midbrain[100],cortex[134],subventricular zone and olfactory bulb[135],single-cell gene expression profiling at whole transcriptome level has not been achieved yet.To integrate spatial information with sequencing,Stahl et al.[136]placed brain sections onto an array with positional barcodes to label transcripts from each location before sequencing.Another technology called Slide-seq[137]coated DNA barcoded beads on slides to mark the spatial position of cells on a tissue section.However,multiple cells can be captured by the same group of arrays or the same bead,making it difficult to guarantee single-cell resolution.Future advancements in spatial transcriptomics profiling platforms will provide a high-resolution brain cell type map and aid novel discoveries in brain connectivity,development,and diseases.
While many studies profile all brain cell types in an unbiased manner, other studies isolate specific cell types by FACS using markers, followed by scRNA-seq, to illustrate the molecular heterogeneity within the population, such as GABAergic neurons [108,115], dopaminergic neurons [116,138,139], microglia [140-143], macrophages [144,145],oligodendrocytes [146], glial progenitors [147], niche cells [119,148], endothelial cells [149], ependymal cells [121], and Drosophila olfactory projection neurons [150]. Moreover,several recent technologies have demonstrated that, by integrating scRNA-seq with other epigenomics, molecular, and cellular features, the functional states of individual cells can be further characterized, leading to better classification and clarification of cell type-specific functions. For example, Lake et al. applied both scRNA-seq and single-cell DNA accessibility assay to the same set of human brain cells for brain cell type classification [105]. Electrophysiological characteristics of single neuron can also be integrated with transcriptome profiling by Patch-seq, thereby elucidating the molecular identity of different excitatory and inhibitory neuron subtypes [151].
Tracking the dynamic transcriptional and genomic landscape in development and aging
While scRNA-seq captures a snapshot of brain cell type compositions in a brain region,it still has limitations in resolving key questions in brain development,including tracing cell lineage,quantifying compositional changes in different developmental stages,and finding connections between cell types during development.Aided by the pseudotemporal analysis algorithms,such as Monocle,Waterfall,and scTDA,the lineage relationships among neurons,stem cells,or even at the whole organism level[34,152,153],can be interpreted from single-cell transcriptome snapshots,reconstructing multiple continuous transition states during development[33,85,86,120,129,154].While these computational pipelines infer trajectories from static landscape of the brain,examining the dynamics in developmental processes through performing scRNA-seq across different time points provides more accurate information and is becoming more popular in in recent studies.
By sampling the brain cell types across multiple time points during embryonic development for scRNA-seq,several studies have addressed the dynamic process of brain development,resolving both cell type heterogeneity,fluctuations and disease associations.Manno et al.characterize the midbrain development by scRNA-seq of human and mouse embryos over time,demonstrating fluctuations in different cell types during development,as well as heterogeneity among dopaminergic neurons,which are known to be associated with Parkinson’s disease[100].Apart from neurogenesis at embryonic stages,at adult stage,the radial glia cells in dentate gyrus of the hippocampus also undergo neurogenesis.By comparing postnatal and adult neurogenesis,similar cell markers and transition stages in development was observed,while their number and spatial distribution differ with age[113].The prefrontal cortex in developing human embryos has also been surveyed using scRNA-seq,presenting the landscape of complex cell types and potential interplays that regulate the balance of excitatory and inhibitory neurons in neural circuits[155].Single-nucleus ATAC-seq of mouse forebrain throughout eight developmental stages also contributed to the identification of cell type complexity,compositional changes and,more importantly,transcriptional regulatory sequences and master regulators that define cell-type identity specification[156].
However,without a cell lineage mark that is stable for accurate lineage tracking,the relationships between progenitors and differentiated cell types are hard to elucidate.To solve this problem,several recent methods utilize CRISPR-Cas9 system to modify endogenous barcode in transgenic zebrafish,demonstrating the plausibility of simultaneous detection of cell lineage and transcriptome information in individual cells in the whole organism[157-159].One of these methods,scGESTALT,utilizes Cas9 to generates random mutations in the lineage barcode at the 3′UTR of DsRed transgene,which is later transcribed with the DsRed mRNA and sequenced with other transcripts in zebrafish brain[157],allowing the simultaneous detection of cell lineage and transcriptome information.While cell lineage tracing at the whole organism level can be achieved in animals with smaller body size,it remains challenging to perform scRNA-seq with lineage tracing in mice.Alternatively,Telly et al.combined the FlashTag system with scRNA-seq to pulse-label progenitor cells in the mouse neo-cortex and trace their daughter cells,and unraveled both intrinsic and extrinsic signals that influence the differentiation and diversification of neurons[160].
In addition to the advancement in understanding cellular programs in early development,scRNA-seq has also provided new insights into the transcriptional changes during aging.Sampling Drosophila whole brain across its lifespan,Davie et al.observed a decline in the RNA content and heterogeneity in gene networks involved in energy consumption in agedbrain,while neuronal identity is minimally affected[161].In mouse ventricular-subventricular zone,infiltration of T cells and a decrease in activated neural stem cells were observed during aging,together with transcriptional changes in endothelial cells and microglia in neurogenic niches[123].Moreover,neural stem cell activity does not decrease during aging,while nichederived inflammatory signals and Wnt antagonist suppresses neural stem cell activation,providing potential therapeutic opportunity in treating neurodegenerative diseases[122].
Apart from dynamics in transcriptional and epigenetic regulations in brain development,the accumulation of somatic mutations at each cell division may also play key roles in producing genomic mosaicism at the whole organism level,resulting in the generation of pathogenic somatic mutations,alterations in local cellular compositions in brain and further effects on the neural circuits.To tackle the brain mosaicism in humans,single-cell whole-genome sequencing of neurons from the same donor can be employed to elucidate all genomic alterations in individual neurons for building a tree model that traces back the history of genome divergence during development.Each neuron was found to harbor~1000 to 1500 single-nucleotide variations(SNVs),which are more frequently located in highly transcribed genes for neuronal functions[162].Sampling neuronal progenitor cells from three fetal human brains,Bae et al.showed the different mutational rates during development,with~1.3 mutations per division per cell at postzygotic cleavages,and increased mutation rate with oxidative damage signature in later developmental stages(including neurogenesis)[163].Comparing young and old individuals(aged from 4 months to 82 years old),the number of somatic SNVs in neurons shows a linear increase in respect to age.Moreover,three different somatic mutation signatures were identified,which correspond to aging process,brain region-specific mutations,and DNA repair in response to oxidative damages.Interestingly,the last signature was also enriched in the neurons from patients affected by early-onset neurodegeneration,including Cockayne syndrome and xeroderma pigmentosum,which are caused by genetic deficits in DNA repair[164].Somatic SNVs,along with copy number variations[165-167]and L1 retrotransposition events[168-170],have been characterized by single-cell whole-genome profi ling,revealing their roles in reshaping the genome of the whole organism throughout the process of development.Thesefindings also shed light on the pattern and frequency of somatic mutations,and further imply that pathogenic somatic mutations can also lead to various neurodevelopmental and neurodegenerative diseases[171,172].
Identifying cell populations associated with neuronal diseases
Neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS), share common pathologies of protein aggregations,synaptic loss, and neuronal death. In recent years, various studies have shed light on potential roles of neuroinflammation in neurodegenerative diseases [173,174].Glial cells, especially microglia, have been shown to maintain brain microenvironment homeostasis and, when reprogrammed in the diseased brain, promote AD progression [175]. However, limited by the number of available cell typespecific markers, the full spectrum of immune cell types and activation states has not been characterized by previous studies.
To achieve a comprehensive unbiased sampling of immune cell populations in brain,Keren-Shaul et al.[176]sampled all immune cells in the brain of wild-type and AD mouse model(5×FAD mice,which expresses five human familial AD gene mutations)using scRNA-seq.A novel type of microglia associated with neurodegenerative disease,disease-associated microglia(DAM),is found to be present only in AD,which results from the gradual deviation from the homeostatic microglia state during disease progression.Characterized by the downregulation of microglia homeostatic factors and induction of lipid metabolism and phagocytic pathways,the DAM represents an activated population of microglia and is involved in plague clearance.The enrichment of DAM in the vicinity of amyloid beta(Aβ)plaques,as well as the observations of increased pool of DAM in AD patients and in an ALS mouse model,suggests a conserved and general response program of microglia towards the aggregated and misfolded proteins generated in neurodegenerative diseases.Similar observations were reported in another AD mouse model,CK-p25[177].Moreover,by collecting and sequencing brain samples from 48 AD patients at different disease stages,Mathys et al.elucidated early and late disease stage-related transcriptional changes in different cell types,as well as gender-associated differences in transcriptome[178].These studies not only have important implications for the development of AD treatment,but also provide a novel method to search for etiology in the neuro-immune axis in other neurodegenerative diseases.
In addition to AD research,scRNA-seq has recently been applied to resolving the cell type relationships and mechanisms of several neuronal diseases.In PD patient iPSC-derived dopaminergic neurons,gene expression changes related to endoplasmic reticulum stress was observed in comparison with dopaminergic neurons from control individuals,and HDAC4 was identified as the upstream regulator of disease progression and potential drug target[139].In multiple sclerosis,lineageand region-specific transcriptomic alterations were also observed,which were associated with cortical neuron damage and glial activation[179].In autism,upper-layer excitatory neurons and microglia were identified as the susceptible cell types affected by the disease[180].Identification of the underlying cell types and regulators of these neuronal diseases would provide new insights into disease mechanisms and opportunities for therapeutic design.
Resolving heterogeneity in brain tumors
Glioma represents the majority of brain tumor in adults.Common genomic alterations in gliomas include mutations in IDH1,TP53,ATRX,and TERT promoter,amplification and rearrangements of EGFR,MET,and PDGFRA,as well as deletions of chromosome 1p/19q and CDKN2A[181,182].
The high intratumoral heterogeneity(ITH),marked by the diversity of genomic alterations,cell lineages,and tumor microenvironment,may be an important reason for the refractoriness of glioma.The ITH in high-grade glioma was elucidated in a scRNA-seq study through profiling gene expression of single cells in EGFR amplified and PDGFRA amplified tumors[183].Muller et al.found that(i)within the same tumor,different cells express distinct EGFR or PDGFRA isoforms;(ii)multiple EGFR oncogenic variants are coexpressed in the same cell;and(iii)some cells express receptor and ligand other than EGFR or PDGFRA.These results suggest that heterogeneity of different tumor clones contributes to the failure of EGFR and PDGFRA inhibitors for glioma treatment.Afterwards,intra-glioma heterogeneity has been repeatedly demonstrated in several studies[184-190].Notably,these single-cell studies highlight that,although the bulk tumor can be classified into three molecular subtypes,individual cells within the same tumor mass commonly exhibit different subtype expression profiles.The extensive ITH is closely related to tumor evolution,drug resistance,and relapse.However,Lee et al.[186]investigated single-cell gene expression in samples from a multi-focal glioblastoma patient and found shared PIK3CA activating mutation and over-expression in tumor masses that were located far apart,highlighting that PI3KCA could be a good candidate for the glioma treatment.A recent study investigated the intra-and inter-tumoral heterogeneity of four subtypes of medulloblastoma,another malignant brain cancer.Complementary to the difference in genomic features,distinct cell populations and developmental trajectories were found among the four medulloblastoma subtypes[191].
Based on spatial and pseudotemporal mapping,scRNA-seq also enables the identification of potential cancer stem cell populations and tracing of developmental lineages,and provides insights into the tumorigenesis.In low-grade glioma,Tirosh et al.found that most cancer cells are differentiated into two glial lineages(oligodendrocyte-like or astrocyte-like cells),while a smaller subset of cells appear undifferentiated and resemble neural stem/progenitor cells[185].They also found that actively cycling cells are enriched among stem/progenitor cells,indicating high proliferation of these cells.Additionally,at the single-cell level,Venteicher et al.[192]showed similar expression profile in two types of low-grade glioma(namely astrocytoma and oligodendroglioma,based on histology),implying shared glial lineages,developmental hierarchies,and cell of origin for these two glioma types.The same hierarchical pattern was reconfirmed in diffuse intrinsic pontine glioma(DIPG),a highly-fatal pediatric glioma.Compared to the less aggressive low-grade glioma,the proportion of undifferentiated,cycling stem/progenitor cells was much higher in DIPG with histone H3 lysine-to-methionine mutations[193].In glioblastoma,cancer stem cells were also identified and were found to recapitulate the developmental hierarchy of normal stem cells[194,195].In a recent study,the model of glioma cell types has been further extended to four transitable cellular states to explain the four gene expression-based subtypes in glioblastoma[196].These studies have shed light on a longstanding debate in gliomagenesis and suggest new therapeutic strategies targeting glioma stem cell populations.Using mouse models,Weng et al.tracked the developmental linage of glioma and captured an intermediate stage named oligodendrocyte-progenitor.These cells are abundant,highly proliferative,and likely to transform to malignant glioma.
They also identified Zfp36l1 as the key gene controlling gliomagenesis[147].
scRNA-seq also aids the comprehensive profiling of the microenvironment of brain tumors.Due to the existence of blood brain barrier,the immune system in brain is largely different from other parts of human body.Microglia,a unique group of brain-resident macrophage,as well as the infiltrated bone marrow-derived macrophages,are very abundant in brain tumor.Microglia and macrophages composite~50%of the tumor core in glioblastoma,and participate in enhancing tumor growth,survival,and dissemination[187].The proportion of infiltrating macrophages increases with glioma grade,and is inversely correlated with response to radiotherapy and survival of high-grade glioma patients[188,189].Single cell sequencing of IDH-mutant astrocytoma and IDH-mutant oligodendroglioma revealed that the abundance of microglia and macrophages accounts for the main difference in expression profile between the two types of clinically distinct low-grade gliomas[192].Similarly,profiling of glioblastoma also revealed that tumor microenvironment differs in glioblastoma subtypes[196].Despite the high similarity between microglia and macrophages,evidence suggests that the infiltrated bone marrow-derived macrophages preferentially express immunosuppressive cytokines and alter the tumor microenvironment[197].Several therapeutic strategies against tumor-associated macrophage are under development and may provide new opportunities for glioma treatment.
Future perspectives
Overall,scRNA-seq has been proved to be a powerful highthroughput tool for resolving individual brain cells,enabling comprehensive and high-resolution cell type determination and novel cell marker identification.The great potential has also been demonstrated in studies of brain development and brain diseases.In our perspectives,three potential directions lead the future studies of brain research using single-cell sequencing-based methods.
Firstly,with the accumulating sequenced single cells as well as the increasing capacity of newly developed technologies,new computational methods to handle the big data are extremely necessary.Droplet-based sequencing platforms,for instance,have produced scRNA-seq datasets encompassing more than half a million of single cells[95,96],challenging the speed and memory efficiency of the state-of-the-art tools.Fortunately,tools such as Seurat[198]and Scanpy[199]emphasize the high efficiency in processing large scRNA-seq datasets.We anticipate that more computational tools are emerging to address this obstacle.Secondly,while numerous studies have addressed the compositional variations in different brain regions and the diversity of heterogeneous cell states,very few attempts have been done to integrate cell types from various studies.Due to the difference in experimental protocols and data processing workflows,results from two different studies are hardly comparable,even if they sequence the same region of the brain or the same type of brain disease.Methodologies and computational frameworks to integrate and comparescRNA-seqdatafrom multipleplatformswillbe beneficial for this purpose.Recently,linked inference of genomic experimental relationships(LIGER)is reported for the integration of multi-omics single-cell sequencing data[200].
Thirdly,single-cell multi-omics,which integrate data from multiple platforms,are also highly important for brain studies.A good example has been set in the classification of retinal bipolar cells[201],which integrated a convergent set of morphological(electron microscopic reconstruction),physiological(calcium imaging),and molecular(scRNA-seq)data.Unbiased,systematic collection of molecular,morphological,physiological,functional,and connectional data will greatly benefit our understanding of the organization and function of the brain.
Overall,while we still know little about the brain,the rapidly developing single-cell sequencing technologies has accumulated big data for future explorations and presented us the single-cell-resolution map of the brain that we have never seen before.Despite problems and challenges present,we expect overwhelming progress in the coming decade.
Competing interests
The authors declare no competing interest.
Acknowledgments
This work was supported by the Research Grants Council(RGC)(Grant No.26102719),Hong Kong Special Administrative Region(SAR),China;the National Natural Science Foundation of China(NSFC)(No.31922088);NSFC-RGC Joint Research Scheme(Grant No.N_HKUST606/17),Hong Kong SAR,China;the Collaborative Research Fund(CRF)(Grant Nos.C6002-17GF and C7065-18GF),Hong Kong SAR,China;the Hong Kong Epigenomics Project(EpiHK);and the Innovation and Technology Commission(ITCPD/17-9,ITS/480/18FP),Hong Kong SAR,China.
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Big Data and the Brain:Peeking at the Future
- GliomaDB:A Web Server for Integrating Glioma Omics Data and Interactive Analysis
- PsyMuKB:An Integrative De Novo Variant Knowledge Base for Developmental Disorders
- Identification of Cognitive Dysfunction in Patients with T2DM Using Whole Brain Functional Connectivity
- Machine Learning to Detect Alzheimer’s Disease from Circulating Non-coding RNAs
- Translational Informatics for Parkinson’s Disease:from Big Biomedical Data to Small Actionable Alterations