Hofmeister离子对水溶液中热响应聚合物的局域环境的影响
2019-01-23薛智敏严传玉赵新辉于东琨牟天成
薛智敏 ,严传玉 ,赵新辉 ,于东琨 ,牟天成 ,*
1北京林业大学材料科学与技术学院,北京 100083
2中国人民大学化学系,北京 100872
1 Introduction
Hofmeister ion effect is a very interesting but elusive phenomenon, the importance of which just begins revealing itself in self assembly1, ion recognition2and protein folding regulation3. However, a lack of mechanistic understanding is limiting its applications. It has been observed long before that salts can significantly affect the solubility of proteins in aqueous solutions. As early as 1888, Hofmeister presented in his studies that salts can greatly alter the solubility of bovine serum albumin and egg white in aqueous solutions4,5. It was shown that some salts such as Na2SO4 can significantly induce the clouding or precipitation of proteins in water, whereas other salts such as NaI did not cause any precipitation even at very high salt concentrations. It was also observed that the addition of MgCl2to solution could not start any clouding even at saturation concentration, while adding KCl caused the clouding. These anions and cations were ordered in the Hofmeister series based on their capability of salting-out/salting-in.The Hofmeister series including the anionic ordering: typically,;and cationic ordering: typically,Li+> Ca2+> Mg2+.
Since then, Hofmeister series has been found to be a recurring phenomenon across the solution chemistry. Myriad solutes including proteins/polypeptides6, polymers7and small molecules8,9, are subject to the Hofmeister ion effects.Moreover, specific ion effects can be used to modulate the lower critical solution temperature (LCST) of macromolecules including thermoresponsive polymers and Elastin-like polypeptides10,11. The specific ion effect was not only found to be on solubility, but also on other properties. For example,specific ion effects were also found to affect the allocation of water at interfaces12. Hence, a thorough understanding of specific ion effects would further inspire advanced applications of smart materials in aqueous environment.
However, there is not a unifying theory for Hofmeister series yet13. There were many attempts to extract a general mechanism. The earliest attempt was made by Hofmeister himself in his serial work, in which he exploited the concept of hydration strength from Arrhenius’s theory of electrolytic dissociation or ionic theory4. Due to limited knowledge about electrolyte solutions at that time, he only ascribed the ordering of ions to water-absorbing effect of salts. The idea of water-absorbing effect was further developed into the two roles of ions in regulating hydrogen bond (HB) network of water:structure making and breaking14. The structure making ions were categorized as ‘kosmotropes’ while the structure breaking ions were referred to as ‘chaotropes’. The kosmotropes can be heavily hydrated and thus can pull the hydrated water from the dissolved solutes, leading to the salting-out phenomena whereas chaotropes play an opposite role. Although this categorization of ions provided a simplistic version of mechanism, the molecular details of Hofmeister ion effects is still at large unclear. For example, the concept of chaotropes barely explains the salting-in effects observed by experiments.Moreover, there is amounting experimental data suggest that the ordering of Hofmeister ions can even be reversed,depending on the specific solutes used15,16. The fact that these two concepts cannot be quantized prevents a well defined physical model from arising. Without a model, the effects of other ions than the known ones cannot be predicted, which means we can only re-edit the series when effects of new ions come up.
In recent decades, there are amounting efforts focused on the molecular interactions between solutes, solvent and ions.Experimental studies are advancing from bulk properties such as viscosity to molecular spectroscopy such as NMR. Also, a considerable share of these efforts was focused on running stimulations for model systems. Meanwhile, molecular stimulation combined with experimental study has proven to be fruitful17. With increasing efforts dedicated to this topic,significant progress has been recently made about how Hofmeister ions interact with solute in water18. It has now been clear that specific ions indeed prefer certain binding sites of protein backbone19. And, there was also competition between water molecule and protein backbone for anions and cations20.
With more and more studies suggesting interactions between ions and solutes play roles in Hofmeister ions effect, the nature of the Hofmeister phenomenon become more or less debatable.Yet, whether the Hofmeister ions effect is a bulk effect that can reach beyond many hydration shells or it is a local effect that specific interactions between ions and solutes play key roles, is not entirely clear. In our previous study, theoretical calculations were carried out in addition to the NMR and Attenuated Total Reflectance Infrared (ATR-IR) measurements21,22. The calculations for electrostatic potential surfaces suggested that there were both negatively and positively charged sites along the protein backbone, which might be the reason for preferred binding sites by ions. And the amount of negative/positive charges on the electrostatic surfaces can explain the competition between water and protein backbone for ions.However, it is not conclusive yet that the specific ion effect is a local effect.
Here, we employed an experiment to study Hofmeister effects on two representative thermoresponsive polymers, one of which is poly(N-isopropylacrylamide) (PNIPAM) while the other is poly(2-ethyl-2-oxazoline) (PEOX). Both of the thermoresponsive polymers are subject to Hofmeister effect and become more and more promising for smart drug delivery systems10,23.
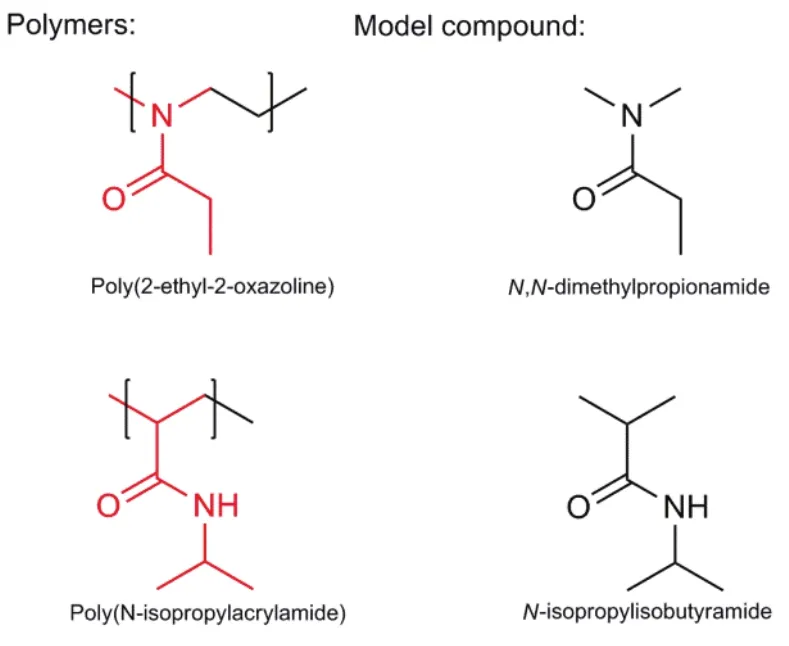
Fig. 1 Molecular structures of two thermoresponsive polymers, poly(2-ethyl-2-oxazoline) and poly (N-isopropylacrylamide) (PNIPAM), and their model compounds: N,N-dimethylpropionamide (NDA) and N-isopropylisobutyramide (NPA), respectively.
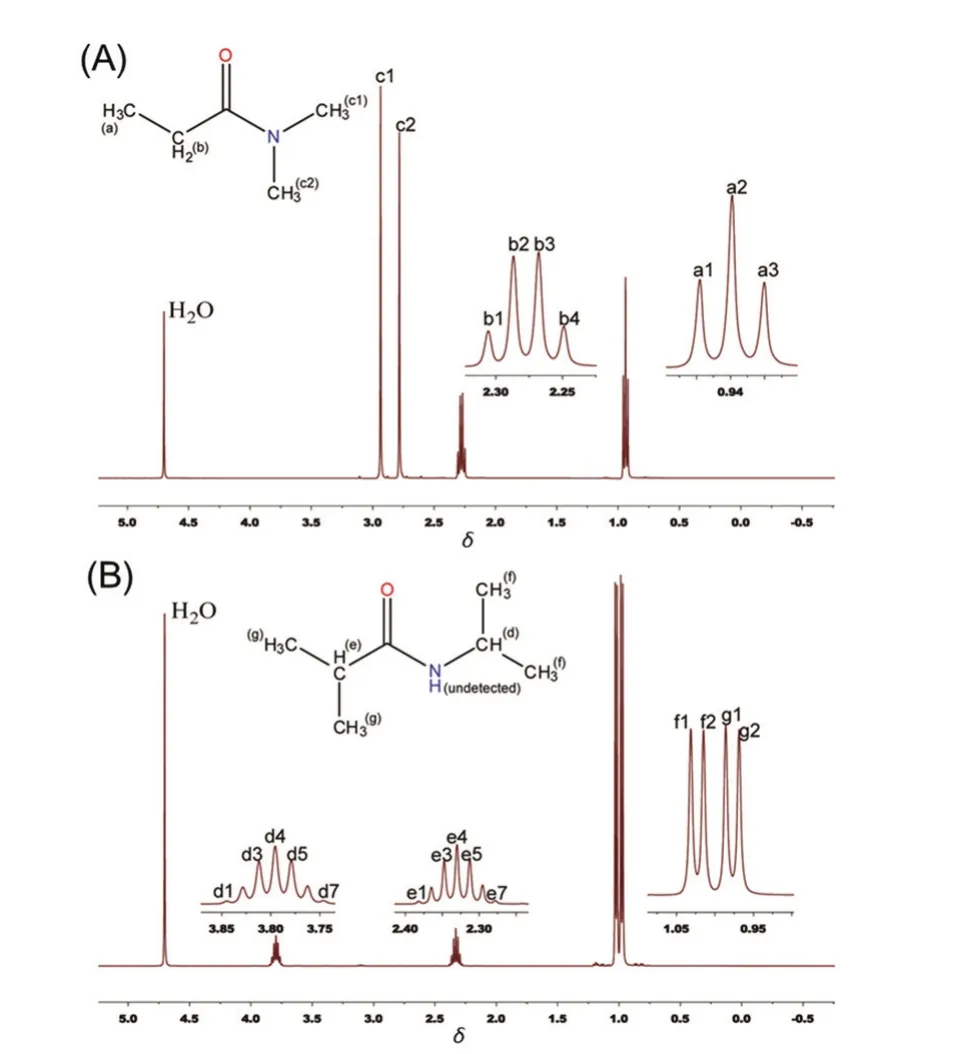
Fig. 2 1H-NMR spectra of model compounds: (A) NDA; (B) NPA.For clarity purpose, the peaks and split-peaks are tagged with alphabetical/-numerical characters.
2 Experimental
All salts (all purities ≥ 99%, except that NaN(CN)2, ≥ 97%and NaBF4, ≥ 98%) and deuterium oxide (D2O, 99.8%) and N,N-dimethylpropionamide (NDA, ≥ 99%) were purchased from J&K Scientific. N-isopropylisobutyramide (NPA, 99.1%)was custom-synthesized by TCI (Shanghai, China). NDA and NPA were used as model compounds of two thermoresponsive polymers (poly (2-ethyl-2-oxazoline) and poly(N-isopropylacrylamide) (PNIPAM)), respectively. Preweighed NDA, NPA and salts were dissolved in D2O to obtain a constant 0.25 mol·L-1for NDA/NPA and various salt concentrations. Details of NMR experiments were given in our previous work21. Briefly,1H-NMR experiments were carried out on a Bruker Advance III 400 HD machine controlled by the Icon NMR program at a constant temperature of 298.15 K.
3 Results and discussion
The phase separation behavior of thermoresponsive polymers PEOX and PNIPAM was subjected to the Hofmeister ion effects. And simple systems consisted of model compounds were usually studied in the search for the molecular mechanism of Hofmeister series. In this case, NDA and NPA were adopted as the model compounds of PEOX and PNIPAM, respectively(Fig. 1). The1H-NMR spectra of model compounds were showed in Fig. 2, and the splitting of the chemical shifts were consistent with their molecular structures. For clarity purpose,the chemical shifts of these compounds were labeled with alphabetical/-numerical characters in order to be easily referred to.
Upon addition of salts, the chemical shifts of NDA were significantly affected by the ionic species (Figs. S1–S4). For example, I-that is a typical chaotropic anion caused the blue shift of chemical shifts (Fig. S1) whereas H2PO4-is kosmotropic and led to the red shift (Fig. S2). Similarly, K+is more kosmotropic than Li+and their effects on the chemical shifts of NDA were de-shielding and slightly shielding,respectively (Figs. S3 and S4).
At increasing salt concentrations, the specific ion effects on chemical shifts were in gradually changing trends (Fig. 3). At the sodium salt concentration of 1 mol·L-1, the changes in chemical shifts for peak a2, b2 and c2 of NDA (Δδa2, Δδb2and Δδc2) range from -0.09 to 0.17, depending on the anion identity. And the ordering of anions for Δδa2was HPO24-<DCA-(dicyanamide anion)This anionic ordering for NDA was almost the same as for N-methylacetamide (NMA)21. Interestingly, the anion effects on Δδb2and Δδc2were almost identical to Δδa2.
Similarly, the range of changes in chemical shift caused by cations was from 0.00 to 0.10 where cationic order for peak Δδa2was this: K+> Na+> GUA+(guanidinium) > N> Ca2+>Li+(Fig. 4A). It was worth noting that the change in chemical shifts was linear after 0.1 mol·L-1, which was consistent with that Hofmeister ion effect is pronounced at ion concentrations above 0.1 mol·L-1. The cation effects on Δδb2and Δδc2were almost identical to Δδa2(Fig. 4B,C).

Fig. 3 Changes in chemical shift as a function of anion concentration for peaks a2, b2 and c2 of NDA (A-C), where the counter cation is Na+.

Fig. 4 Changes in chemical shift as a function of cation concentration for peaks a2, b2 and c2 of NDA (A-C), where the counter anion is Cl-.
Moreover, we also explored how differently ions change different groups on the NDA molecule. Therefore, Δδb2 was plotted against Δδc2(Fig. 5), in which the slope factor ‘k’ was defined as Δδb2/Δδc2. “k = 1.0” means the chemical shifts of proton b and proton c were equally affected by ions. It was found that k was very close to 1.0 for all anion effects (Fig.5A). So were the cationic effects (Fig. 5B). The k value of ~1.0 for both anion and cation effects suggested the Hofmeister ion effect was isotropic on the NDA and therefore it was a global effect, which further implied there were no significant specific interactions between ions and NDA.

Fig. 5 Correlations between Δδb2 and Δδc2: (A) anionic effects where the counter cation is Na+; (B) cationic effects where the counter anion is Cl-.The dash line represents a linear correlation (k is the slope of the dash line, k = Δδb2/Δδc2).

Fig. 6 Changes in chemical shift as a function of ion concentration for peaks d4, e4, f1 and g2 of NPA (A-D):anionic effects where the counter cation is Na+.
Same experiments were carried out for studying specific ion effects on NPA, which was one of model compounds for PNIPAM (Fig. 6). It was worth noting that the solubility of NPA was very sensitive to addition of strong kosmotropic salts such as NaOH at 298.15 K, which led to the phase separation of NPA from solutions even at very low salt concentration. This sensitivity of NPA was consistent with the dramatic decreasing LCST of PNIPAM upon addition of kosmotropic salts even atlow concentrations. Therefore, the effects of most of the kosmotropic anions and K+on NPA were not studied here.
Nevertheless, the changes in chemical shift of d proton of NPA (Δδd4) were found to follow the ordering of anions: PF6-~For the proton e (Δδe4) that is adjacent to the carbonyl group of NPA, the ordering of anions was different as follows: I->6B). And for the proton f (Δδf1) that is also adjacent to the carbonyl group of NPA, the ordering of anions was same as that for Δδe4(Fig. 6C). Interestingly, the anionic ordering for the proton g (Δδg2) that is on nitrogen side of amide bond was same as that for Δδd4(Fig. 6D). However, the cationic ordering for Δδd4, Δδe4, Δδf1and Δδg2was almost the same as follow:Na+> GUA+> NH4+> Li+> Ca2+(Fig. 7).
Compared to NDA, NPA has a larger volume and the local chemical environments and thereby ions distribution around it could possibly be different, namely anisotropicity. In order to study the anisotropicity, the changes in the chemical shifts for different methyl groups oncarbonyl side (Δδd4and Δδf1) were plotted against the changes in the chemical shifts for different methyl groups on carbonyl side (Δδe4and Δδg2) (Fig. 8).Similar to earlier discussions on the isotropicity of ion effects on NDA, the anisotropicity for NPA was also measured by k values, where k was defined as Δδx/Δδy(x = d4 or f1, y = e4 or g2). It was found that correlation between Δδd4to Δδe4was most deflected from isotropic k = 1.0, for both anionic and cationic effects (Fig. 8A,C). However, for Δδd4and Δδe4that represent the protons further away from the amide core of NPA molecule, k values were very close to 1.0 for both anions and cations (Fig. 8B,D).
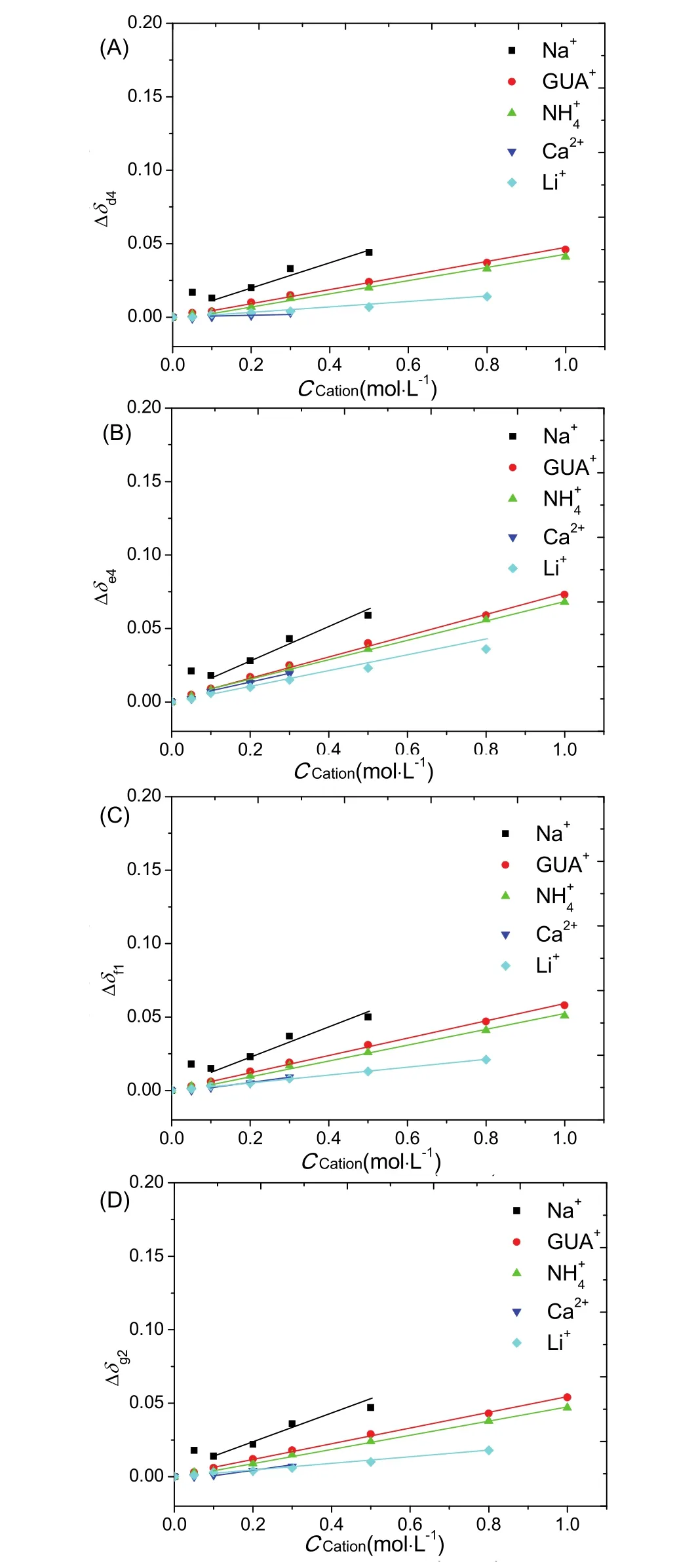
Fig. 7 Changes in chemical shift as a function of ion concentration for peaks d4, e4, f1 and g2 of NPA (A-D): cationic effects where the counter anion is Cl-.
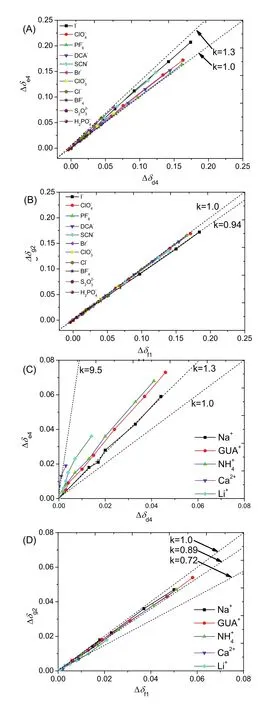
Fig. 8 Correlations between d4 and e4, and correlations between f1 and g2: (A, B) anionic effects where the counter cation is Na+; (C, D) cationic effects where the counter anion is Cl-.The dash line represents a linear correlation (k is the slope of the dash line).
In Fig. 8A, it was found that smaller anions such as halides caused greater anisotropicity between Δδd4and Δδe4, whereas this anion size related anisotropicity was less significant for Δδf1and Δδg2(Fig. 8B). For the cationic effects, it was found that ions of higher charge density such Li+and Ca2+caused greater anisotropicity between Δδd4 and Δδe4 (Fig. 8C), and the anisotropicity was less significant for Δδf1and Δδg2(Fig. 8D).In Fig. 8C, it was found that k value between Δδd4and Δδe4was about 9.5, which suggested that the strength of cationic effects on the carbonyl side of amide bond was about 9.5 times greater than the amide nitrogen side.
In order to find out why the Hofmeister ion effects on NPA were more anisotropic than on NDA, and cationic effects were more anisotropic than anionic effects on NPA, the electrostatic potential surfaces of NDA and NPA were calculated (Fig. 9).The electrostatic potential surface is a useful index of non-covalent interaction. It was found that all protons of NDA were more exposed than the protons of NPA, especially proton d and e on NPA. It was also found that the carbonyl oxygen on NDA (site @1) was less negatively charged than @2, and @3 was less positively charged than @4. Therefore, the fact that specific ion effects were more anisotropic on NPA than on NDA might be due to steric hindrance. And the fact that cationic effects were more anisotropic than anionic effects on NPA might be due to that the interactions between cations and NPA were stronger than the interactions between anions and NPA, for which reason the highly charged cations such as Ca2+caused a significant anisotropicity (Fig. 8C).
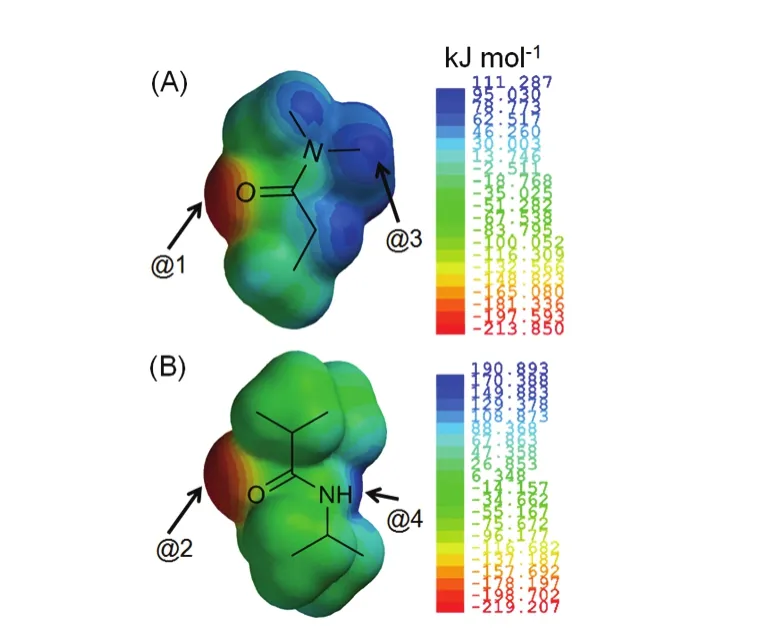
Fig. 9 Electrostatic potential surfaces of model compounds:(A) NDA and (B) NPA. The surfaces were calculated by using DFT at the B3LYP/6-311+G** level with Spartan program.Potential values are in kJ·mol-1 and have a precision of 0.002 IsoValue.@1 and @2 are the negatively charged sites whereas @3 and @4 are the positively charged sites on the electrostatic surfaces.
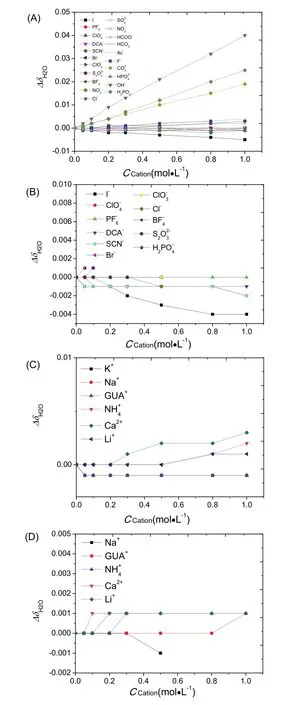
Fig. 10 Changes in chemical shift of water residue (H2O):(A, B) anionic effects where the counter-cation is Na+; (C, D)cationic effects where the counter-anion is Cl-;(A, C) NDA solutions; (B, D) NPA solutions. The solid lines are only guidance to eye.
In addition to the methyl groups of NDA and NPA, the changes in chemical shift of water residue (H2O) in D2O were also plotted against the ionic concentrations (Fig. 10). Overall,the chemical shift of water proton was shifted to lower field(ΔδH2O> 0) after additions of strong kosmotropes such as OH-and H2P, whereas increasing concentration of strong chaotropes such as I-and SCN-led to high-field shifting(ΔδH2O< 0) but to a less extent (Fig. 10A,B). Between strong water proton was as minimal (ΔδH2O~ 0). Nonetheless, the ordering of anions for water was this: OH-> HPO42-~Li+> GUA+~ Na+~ K+(Fig. 10C,D).
The results in Fig. 10 provided a quantitative explanation to these two terms. For example, the ΔδH2Oof conventional kosmotropic anions (including OH-, HPO42-, H2PO4-, CO32-,Ac-, SO42-, F-, S2O32-, HCO3-and HCOO-) was > 0.00, which suggested that the stronger HB and more charge transfer between water and these anions. And the ΔδH2Oof extremely strong kosmotropic anions such as OH-was >> 0.00, which supported that OH-was a very strong HB acceptor. And the ΔδH2O of conventional chaotropic anions (such as I-and SCN-)was < 0.00. Surprisingly, the ΔδH2Oof Cl-at concentration lower than 1 mol·L-1was 0.00, demonstrating to be the benchmark between chaotropes and kosmotropes. Despite of different solutes, the results were indifferent in both NDA and NPA solutions. These results suggested a quantitative measure of kosmotropicity/chaotropicity that the anion would be kosmotropic if its ΔδH2Owas higher than that of Cl-and vice versa. Moreover, the results showed that the effects of cations on water structure were minimal, which was consistent with that there was minimal charge transfer between cations and water24.

Fig. 11 Electrostatic interactions between solutes and ions: anions(left) and cations (right).Due to repulsive forces between nuclei, there is an equilibrium distance r0 (or r1)between anions (or cation) and solute molecules. For a given combination of ion and solute, the value of r is fixed.

Fig. 12 Cation-induced resonance structures of amide bonds.Cations of relatively low charge density such as Na+ are likely to shift the amide bond to a planar one (A), whereas cations of higher charge density such as Ca2+ are more likely to polarize the carbonyl group (B).
As a rule of thumb, small particles are always in non-stopping movements so that the influence of ions on solute molecules is a statistically average result. That explained the close Δδ (k = 1.0) upon addition of salts for most of methyl protons (Fig. 5A,B). However, this is under the assumption that the size of solute molecules can fit into the electrostatic field of ions (upside, electrostatic-charged circles in Fig. 11). It was worth mentioning that it was negative charges accumulated on the surface of solute molecules under influence of cations although the Δδ for all cations were >> 0.00. Since the weak effects of cations were offset by the strong effects of Cl-, the surfaces of solutes were in whole positively-charged and thus the Δδ for all cations were >> 0.00 (Figs. 4A–C and 7A–D).
In the case of NDA, the volume of ions is close to that of solute (Table S1). However, the volume of NPA is 30% larger than that of NDA. Thus, the size in volume of some ions is quite small in comparison to the size of NPA, which would lead to the uneven distribution of charge on the surface of solute because the solute cannot wholly fitted into the electric field of ions (downside, electrostatic-charged circles in Fig.11). That explained the non-proportional relationships between Δδ (k > 1.0 or k < 1.0) (Fig. 8A–D). For example, for big-sized anions such as PF6-, the slope rate ‘k’ for Δδe as a function of Δδbwas equal to 1.0; whereas, for smaller-sized anions of Cl-,Br–and I–, k = 1.3 (Fig. 8A).
The volume effects can explain the slight disproportion between most of Δδ. However, there must be other reasons for the splitting of Δδc1from Δδc2and dramatic differences between Δδe from Δδb under influence of cations (Fig. 8C). A highly possible explanation for that would be the increasing percentage of one of three resonance structures of amide bond.Under certain conditions, one of three resonance structures would dominativ25,26. And it could be that cations of relatively low charge density such as Na+are likely to shift the amide bond to a planar one (Fig. 12A), whereas cations of higher charge density such as Ca2+are more likely to polarize the carbonyl group (Fig. 12B). As a result of the planarization of amide bond, the volume effects would be more significant for small anions such as I-and NAnd due to the polarization of carbonyl group, the differences between Δδefrom Δδbwould be more dramatic under influence of highly-charged cations such as Ca2+(Fig. 8C). Since there were two isopropyl groups adjacent to amide bond in NPA, the carbonyl group of NPA is more likely to be polarized than that of NDA is. Thus, the differences between Δδb2 from Δδc2 were less prominent under influence of cations (Fig. 12).
4 Conclusions
In summary, we employed nuclear magnetic resonance spectroscopy to investigate the specific ion effects on the N,N-dimethylpropionamide and N-isopropylisobutyramide,which are the model compounds of poly(2-ethyl-2-oxazoline)and poly(N-isopropylacrylamide), respectively. It was found that the Hofmeister ion effects on the model compounds of the two polymers followed the same ion orderings of our previous study, which suggested that Hofmeister ion effect is irrespective of solutes and thus it might be a global effect. The anisotropicity between different methyl groups suggested that local interactions between ions and model compounds also play a role. Moreover, the change in chemical shift of δH2Ocould be a proof of concept of water structure making and breaking for kosmotropes and chaotropes.
Supporting Information:available free of charge via the internet at http://www.whxb.pku.edu.cn.
