蓬乱蛋白2干扰和过表达慢病毒载体的构建及其在hUVECs中的表达*
2019-01-18生欣王俊华刘月华郜双林谢夏丹范芳
生欣,王俊华,刘月华,郜双林,谢夏丹,范芳
(遵义医学院基础医学院 生物化学教研室,贵州 遵义 563000)
蓬乱蛋白(Dishevelled, DVL)是一类穿梭于胞质和细胞核的Wnt信号通路蛋白,能够与多种蛋白相互作用,将Wnt信号由膜受体Frizzled传递至下游组件[1]。DVL最早发现于果蝇体毛和翅毛异常定位表型中,在进化上高度保守[2]。目前在人类中已知的DVL有3个亚型(DVL1,DVL2和DVL3),均具有3个高度保守的结构域,即N-端的DIX结构域、内部的PDZ结构域和C-端的DEP结构域[3-4]。该蛋白不仅参与细胞的增殖、迁移和凋亡,而且与胚胎发育、细胞极性、分化及肿瘤发生等过程相关[5-6]。尽管通过结构域特征能够预测其功能,但是该蛋白在不同组织和细胞中确切的生物学功能还不清楚[7]。
近年来,大量研究显示Wnt信号通路参与动脉粥样硬化的发生[8-9]。Wnt5α能够通过经典Wnt信号通路引发内皮细胞炎症反应[10-11],且该通路过度激活能够导致平滑肌细胞增殖和血管内膜增厚[12-13],但有关DVL在该过程中的作用还未见报道。本课题组在动脉粥样硬化的体内外模型中观察到DVL2的异常表达,然而,其具体的作用机制还不清楚。相比常规的逆转录病毒和腺病毒表达系统,慢病毒表达系统能够将病毒基因组整合到宿主基因组,从而实现长时间稳定表达,且具有能够感染非分裂期细胞、产生免疫反应小、可插入大片段外源基因等优点,成为导入外源基因的有利工具,并被广泛应用于各种细胞系、原代细胞和干细胞的基因过表达、RNA干扰、microRNA研究,以及基因缺陷型动物模型复制等研究中[14]。因此,本研究拟构建DVL2的慢病毒干扰和过表达载体,建立人脐静脉内皮细胞(human umbilical vein endothelial cells,hUVECs)的稳定表达株,并在体外研究其对DVL2基因表达的影响,以期为进一步探索DVL2在动脉粥样硬化发生早期的作用及其蛋白互作关系奠定实验基础。
1 材料与方法
1.1 材料
实验所用的慢病毒系统pHBLV-CMV-MCS-3flag-EF1-ZsGreen-puro、pHB-U6-CMV-MCS-PGKZsGreen-puro、感受态DH5α细胞均由遵义医学院基础医学院生物化学教研室保存,hUVECs、HEK 293T细胞株和含有生长因子的内皮细胞专用培养基购自美国典型培养物保藏中心(ATCC),pSPAX2、pMD2G(美国Addgene公司),转染试剂Lipofectamine 2000、DMSO、限制性内切酶EcoRⅠ和BamHⅠ、T4连接酶、质粒提取与纯化试剂盒PureLinkTMHiPure Plasmid Maxiprep Kit、DNA Polymerease、DNA ladder、 蛋 白Marker(美国Thermo Fisher Scientific公司),载体回收试剂盒MiniBEST Agarose Gel DNA Extraction Kit Ver.4.0(日本TaKaRa公司),凝胶回收试剂盒、BCA定量试剂盒(北京天根生化科技有限公司),PCR引物、琼脂糖、琼脂粉(上海生工生物工程股份有限公司)。
1.2 方法
1.2.1 目的基因h-DVL2CDS区域的扩增 提取hUVECs细胞基因组DNA,设计正向引物h-DVL2-Eco/Eco-F(5'-GGATCTATTTCCGGTGAATTCGCCACCATG GCGGGTAGCAGCACTG-3')和反向引物h-DVL2-Eco/Eco-R(5'-TCTAGAACTAGTCTCGAGCATAACATCCA CAAAGAACTCGCT-3')。以基因组DNA为模板扩增合成目的基因h-DVL2的CDS区域,反应条件为:95℃预变性 5 min,94℃变性20 s,55℃退火 20 s,72℃ 延伸150 s,共27个循环,72℃继续延伸10 min;PCR产物电泳回收纯化,测序分析。
1.2.2 干扰靶点设计和引物合成 以人DVL2基因的mRNA序列(GeneBank Accession No.NM_004422.2)根据shRNA设计原则设计shRNA靶序列和对照病毒载体(见表1、2)。合成引物,分别稀释至100 μmol,引物退火(95℃ 10 min,75℃ 10 min,55℃ 10 min,35℃ 10 min,15℃ 10 min)形成带黏性末端的双链片段,退火处理后得到shRNA模板用于连接反应。
1.2.3 过表达和干扰表达载体的构建 分别用限制性内切酶EcoRⅠ和BamHⅠ酶切目的基因和慢病毒载体,将目的基因与线性化慢病毒过表达载体pHBLVCMV-MCS-3flag-EF1-ZsGreen-puro连接,插入MCS区,获得重组过表达质粒pHBLV-HDVL2。将shRNA模板与线性化的慢病毒干扰载体pHB-U6-CMVMCS-PGK-ZsGreen-puro连接,插入U6启动子后,获得重组干扰质粒pHB-shRNA-HDVL2。分别转化到大肠杆菌DH5α中,通过菌液PCR鉴定,将阳性克隆菌液测序分析。
1.2.4 重组表达h-DVL2慢病毒在HEK293T细胞中的包装和转染 采用3质粒系统进行病毒包装,包括慢病毒穿梭质粒,即已经制备的重组慢病毒质粒pHBLV-HDVL2/pHB-shRNA-HDVL2,2种辅助包装原件质粒,即pSPAX2和pMD2G。采用高纯度无内毒素抽提3种质粒载体,浓度达到1 μg/μl,A260/280在1.7~1.8用于病毒包装。以Lipofectamine 2000脂质体同时将3质粒系统转染HEK293T细胞,转染后6 h更换含10% FBS的新鲜完全培养基。在48 h时,收集1次培养基,补入新鲜完全培养基后继续培养,在72 h收集所有培养基,离心去除细胞碎片,收集病毒原液于超速离心管中,4℃、22 206 r/min离心120 min,分装病毒超离液,置入-80℃冰箱冷冻保存。

表1 靶向DVL2基因的shRNA寡核苷酸单链
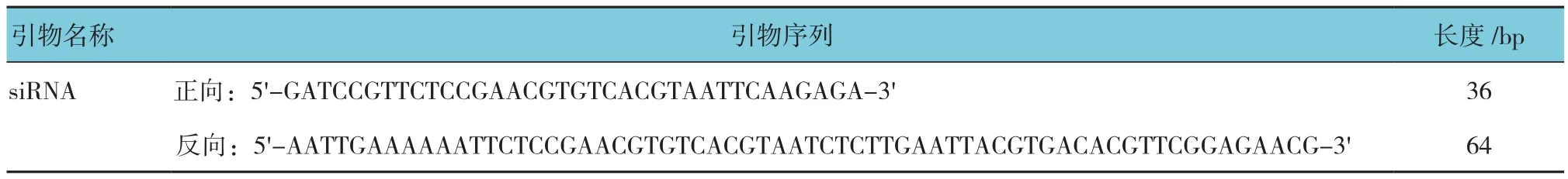
表2 对照病毒载体siRNA寡核苷酸单链
1.2.5 病毒滴度测定 HEK293T细胞以1×105个/ml的密度种96孔板,100 μl/孔,每个病毒准备6孔,37℃,5%二氧化碳CO2培养箱中培养过夜。将10 μl病毒液做3倍梯度稀释,共6个稀释度,分别加到对应的孔中,并追加培养基至100 μl;48 h后,换液为100 μl含1.5 μg/ml嘌呤霉素的完全培养基;24 h后,换完全培养基继续培养6 h,活细胞工作站观察荧光,荧光百分比在10%~30%的孔计算病毒滴度;滴度(TU/ml)=细胞数×荧光百分比×MOI×病毒稀释倍数×103。
1.2.6 慢病毒感染hUVECs 以1×105个/ml的密度接种hUVECs到6孔板,细胞融合度在50%~70%更换含有聚凝胺(终浓度为6 μg/ml)的适应性培养基,根据病毒滴度和细胞量加入相应体积的病毒原液;感染后24 h,换无病毒的适应性培养基,37℃继续培养24 h后,活细胞工作站观察,绝大部分细胞感染病毒以后,利用嘌呤霉素抗性筛选掉未感染病毒的细胞;换嘌呤霉素浓度为8 μg/ml的适应性培养基,37℃培养1周,直至没有感染病毒的细胞被嘌呤霉素杀光,将嘌呤霉素浓度降低到4 μg/ml继续培养1周,使细胞慢病毒感染稳定传代;由于感染效率高,稳定性好,故不需要进行挑取单克隆操作,将细胞扩大培养后即可正常使用。将感染好的细胞分为BC组(hUVECs空白对照)、NC组(HBLV-GFP-PURO阴性对照)、干扰组(pHB-shRNA-HDVL2)及过表达组(pHBLVHDVL2)。
1.2.7 实时荧光定量聚合酶链反应(qRT-PCR)检测mRNA相对表达量 参照试剂盒说明书,分别以RNAiso Plus提取总RNA,以Prime ScriptTMRT Master Mix进行逆转录,逆转录条件为:37℃预反应15 min;85℃扩增5 s。引物序列见表3。参照TB GreenTMPremix Ex TaqTMⅡ(Tli RNaseH Plus)试剂盒说明书,进行qRT-PCR反应,反应条件为:95℃预变性30 s;95℃变性5 s,60℃ 退火30 s。共40个循环。数据以2-ΔΔCt进行计算。
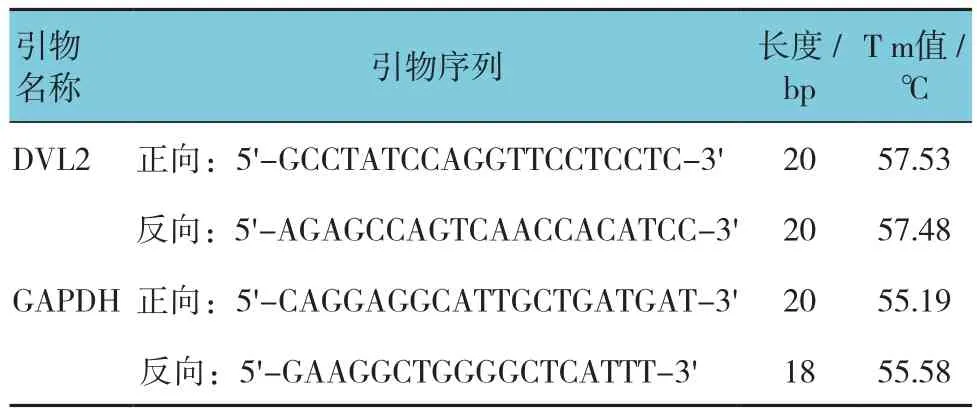
表3 qRT-PCR引物序列
1.2.8 Western blotting检测蛋白的相对表达量 细胞消化裂解,提取蛋白,以BCA法进行蛋白定量,以5×蛋白Loading buffer按4∶1充分混合,100℃煮沸10 min变性蛋白。每孔按BC组、NC组、干扰组、过表达组顺序各上25 μg蛋白样品溶液。80 V恒压电泳,200 mA电转120 min;5%脱脂奶粉室温封闭2 h,1×TBST,清洗6次,每次5 min;分别以兔抗人DVL2(1∶1 000)和小鼠抗人GAPDH一抗(1∶10 000)4℃,过夜孵育,1×TBST清洗;分别以山羊抗兔和兔抗小鼠二抗(1∶3 000),室温孵育2 h,1×TBST清洗;以Chemi DocTMtouch imaging System(BIO-RAD)检测蛋白条带ECL化学发光显色,以ImageLab软件检测蛋白条带灰度值,目的蛋白的相对含量=目的条带灰度值/对应GAPDH灰度值。
1.3 统计学方法
数据分析采用SPSS 19.0统计软件,计量资料以均数±标准差(±s)表示,多样本比较采用单因素方差分析,其两两比较采用LSD-t检验,P<0.05为差异有统计学意义。每组实验均重复3次,每个样本每次检测做3个复孔。
2 结果
2.1 pHBLV-HDVL2过表达慢病毒载体鉴定结果
将转化后的阳性克隆菌液进行PCR扩增,1%琼脂糖凝胶电泳鉴定,可见整齐均一的条带(见图1A~D),对照DNA Marker进一步确定阳性克隆,菌液送检进行测序鉴定,结果显示插入序列与构建的目的片段碱基序列完全一致(见图1E),质粒构建成功。
2.2 h-DVL2 shRNA慢病毒载体鉴定结果
将构建的腺病毒干扰载体pHB-shRNA-HDVL2酶切,1%琼脂糖凝胶电泳鉴定(见图2A、B),胶回收。将重组的阳性克隆进行PCR扩增,得单一产物,将产物进行测序,测序结果见图2C。测序峰图(见图2D)显示在测序结果的发夹结构中,干扰序列正确。其干扰序列峰形图均为单峰,无突变pHB-shRNA-HDVL2构建成功。
2.3 慢病毒载体的包装及滴度测定结果
慢病毒载体转染HEK293T细胞后,荧光百分比在10%~30%的孔计算病毒滴度(见图3),NC组、干扰组、过表达组滴度分别为2×108、2×108和1×108TU/ml。
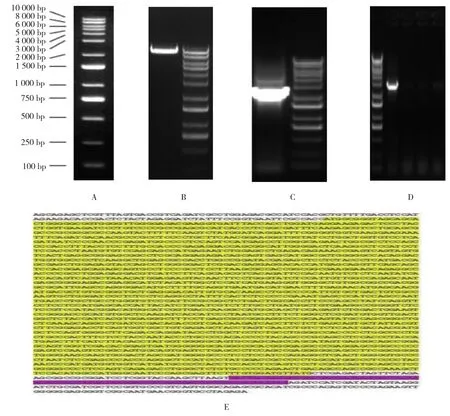
图1 pHBLV-HDVL2过表达载体的构建和测序结果
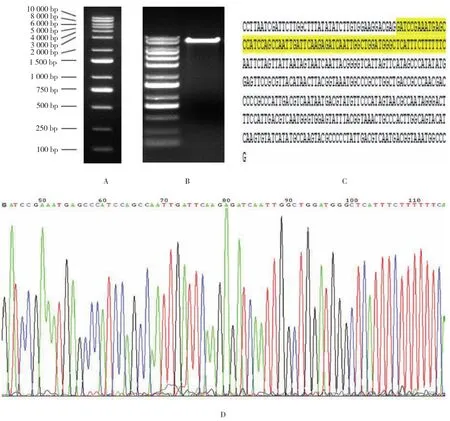
图2 pHB-shRNA-HDVL2 shRNA干扰表达载体的构建和测序结果
2.4 慢病毒感染hUVECs的感染率
慢病毒感染hUVECs后,利用puro+抗性基因,加puromycin筛选掉未感染细胞。通过Image J分析得感染率>98%(感染率=带荧光细胞数/细胞总数)(见图 4)。
2.5 DVL2 mRNA和蛋白相对表达量比较
各组DVL2 mRNA相对表达量比较,经单因素方差分析,差异有统计学意义(P<0.05);进一步两两比较显示,NC组中DVL2 mRNA相对表达量与BC组比较,差异无统计学意义(t=1.910,P=0.129);干扰组DVL2 mRNA相对表达量较BC组降低(t=28.369,P=0.000),沉默效率为67%;过表达组中DVL2 mRNA相对表达量增加(t=-5.641,P=0.005),过表达水平为NC组的5.6倍。
各组DVL2蛋白表达比较,经单因素方差分析,差异有统计学意义(P<0.05);BC和NC组中可见DVL2条带较细,而过表达组较粗。进一步两两比较显示,NC组中DVL2蛋白表达较BC组差异无统计学意义(t=0.031,P=0.977);干扰组DVL2灰度较BC组降低(t=37.025,P=0.000),干扰效率为61%;过表达组中DVL2蛋白表达增加(t=-13.004,P=0.000),表达水平为BC组的2.7倍。见图5和表4。
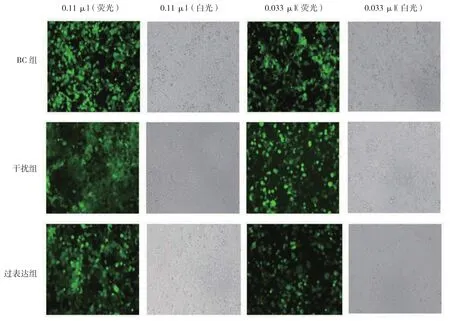
图3 慢病毒滴度测定图 (×100)
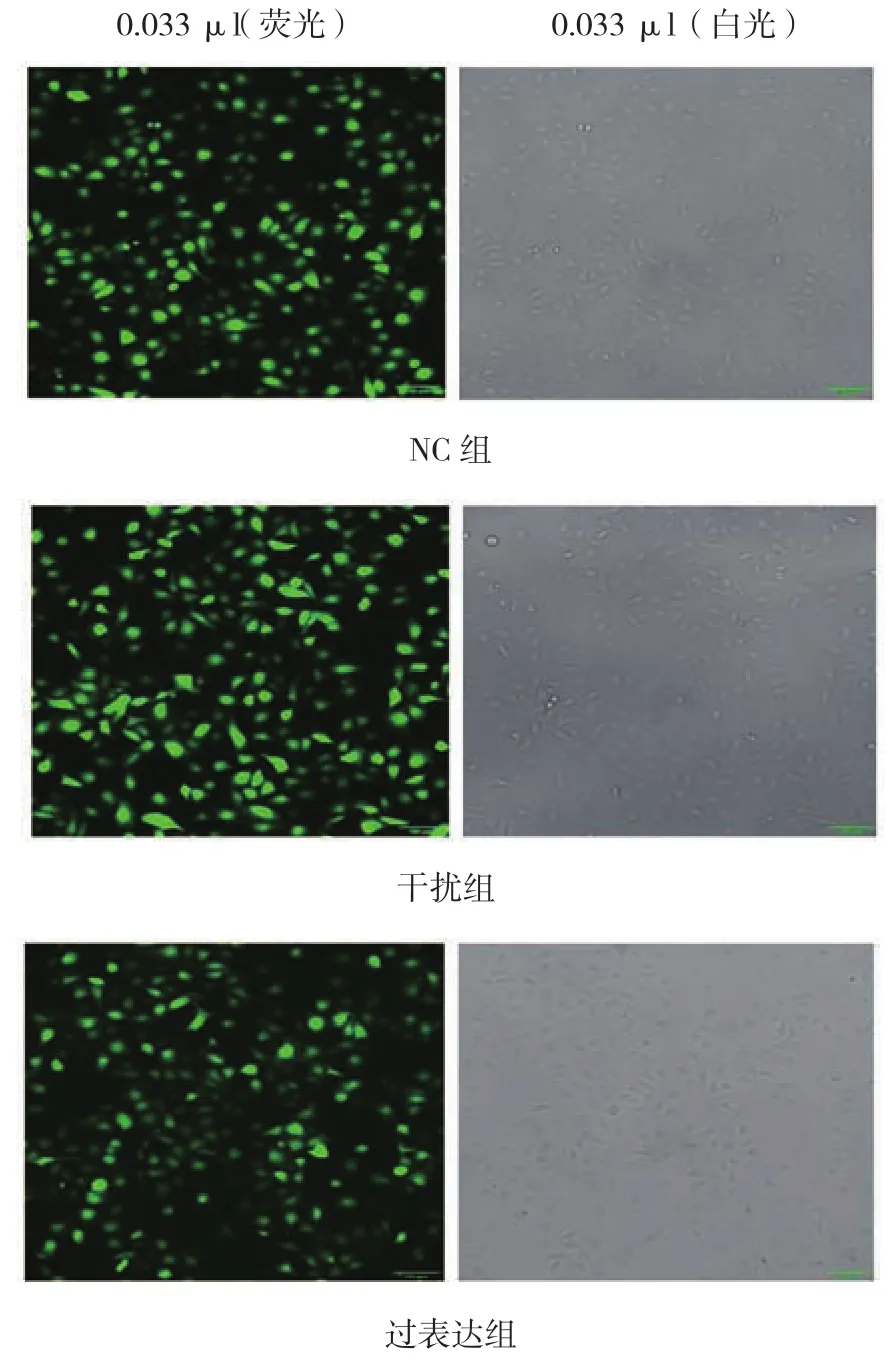
图4 慢病毒感染hUVECs荧光鉴定图 (×100)
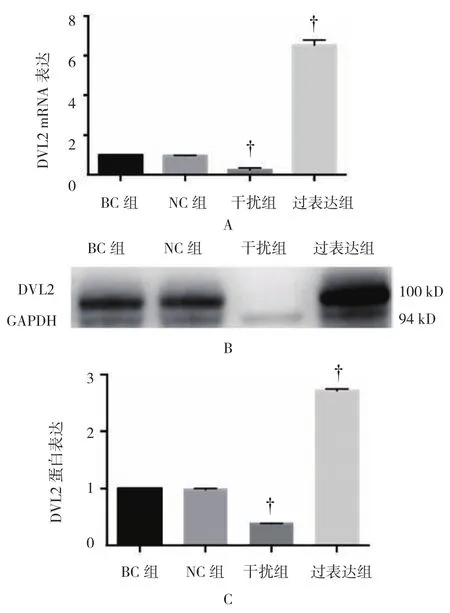
图5 各组DVL2 mRNA和蛋白表达比较
表4 DVL2 mRNA和蛋白表达比较(n =3,±s)
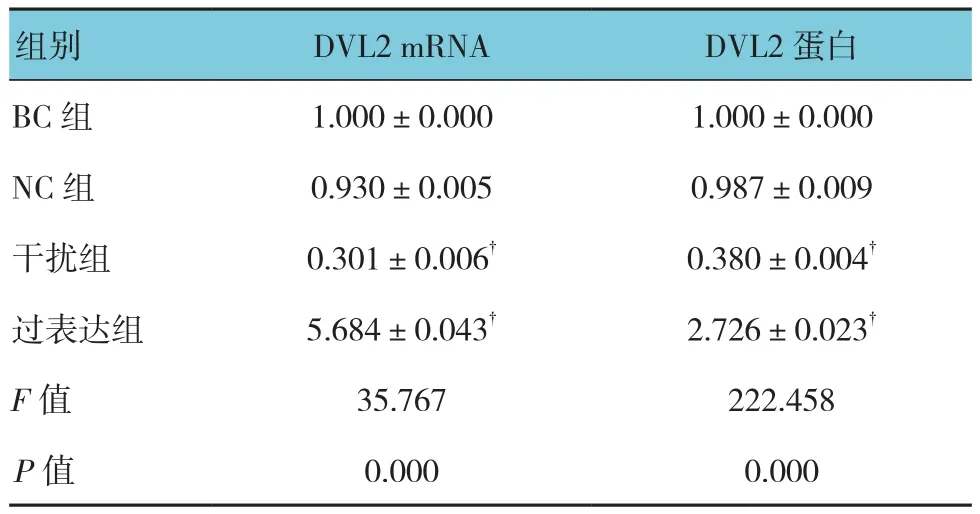
表4 DVL2 mRNA和蛋白表达比较(n =3,±s)
注:†与BC组比较,P <0.05
组别 DVL2 mRNA DVL2蛋白BC 组 1.000±0.000 1.000±0.000 NC 组 0.930±0.005 0.987±0.009干扰组 0.301±0.006† 0.380±0.004†过表达组 5.684±0.043† 2.726±0.023†F值 35.767 222.458 P值 0.000 0.000
3 讨论
本研究首先构建DVL2过表达和干扰慢病毒表达载体pHBLV-HDVL2和pHB-shRNA-HDVL2,即穿梭质粒,酶切和测序结果显示质粒构建成功。进一步采用3质粒系统,将表达载体和包装质粒pSPAX2和pMD2G共转染至HEK293T细胞中,进行病毒包装形成具有感染能力的慢病毒颗粒。由于表达载体携带有ZsGreen绿色荧光蛋白基因,较常规的EGFP荧光更为稳定,通过荧光显微镜观察能够获得病毒包装的滴度和转染效率。原代细胞为较难转染的细胞,本研究通过慢病毒表达系统,能够将携带DVL2的过表达和干扰shRNA序列整合到原代hUVECs基因组中,并通过puromycin筛选出稳定表达的细胞株,获得较高的转染效率(>98%)。与BC组比较,过表达组的DVL2表达增加,而干扰组的DVL2表达水平下调。可见,本研究成功获得了DVL2的干扰和过表达慢病毒载体,并能够在原代hUVECs细胞中稳定表达。
Wnt信号通路是由分泌型糖蛋白Wnt激活的一系列信号通路,包括经典的Wnt/β-caternin信号通路,非经典的Wnt/PCP信号通路和Wnt/Ca2+信号通路[15]。Wnt信号通路与动脉粥样硬化密切相关。在临床报道中[16],经典Wnt信号通路受体低密度受体脂蛋白相关蛋白6(LRP6)基因突变患者伴随动脉粥样硬化的风险更高,而LDLR-/-小鼠模型已经成为研究动脉粥样硬化的模式动物。经典Wnt信号通路配体Wnt 5α[17],下游蛋白β-caternin[18]及其通路抑制剂DKK-1[19]均参与动脉粥样硬化的发生和动脉粥样斑块的维持。因此Wnt/LRP6/β-catenin信号通路活性的改变已经成为动脉粥样硬化斑块形成的一大原因。而有报道显示,非经典Wnt/PCP信号通路同样也与动脉粥样硬化相关,其下游RoA/ROCK也参与动脉粥样硬化早期的内皮细胞功能损伤和后期的斑块形成和破裂[20]。DVL作为Wnt信号通路的关键蛋白,同时参与多条Wnt信号通路,是一个关键的枢纽分子[1]。在不同的Wnt信号通路中分别与不同的蛋白相互作用调控不同细胞活动。尽管Wnt信号通路与动脉粥样硬化的关系已经得到广泛的认可[21-22],然而,有关DVL在动脉粥样硬化过程中的作用机制还清楚,只有少量研究显示DKK通过激活DVL1参与血管内皮细胞迁移[23-24],也有研究猜测DVL可能通过Wnt/PCP信号通路改变血管内皮细胞的平面极性[25]。然而,DVL在动脉粥样硬化过程中通过与哪一些蛋白质结合,以及通过哪一条Wnt信号通路调控内皮细胞功能,并进一步参与动脉粥样硬化的发生还不清楚。此外,相比DVL1和DVL3,DVL2的功能研究还较为缺乏,因此,成功构建DVL2干扰和过表达载体对于深入研究DVL2在动脉粥样硬化发生过程中的作用及其所涉及的Wnt通路蛋白具有重要的意义。
