用三重四级杆气质联用仪测定水产品中多种麻醉剂
2019-01-12,,,
,,,
(1.苏州检验检疫局综合技术中心,江苏 苏州 215104; 2.苏州华博日化品检测服务有限公司,江苏 苏州 215104)
由于水产品种分布的多样性, 鲜活水产品跨地区、长时间运输已成为常态[1]。因此,在长途运输中对水产品进行有效保活就显得非常重要[2],在水产品运输中使用麻醉剂可以降低鱼类的耗氧量和氨氮排放量,可有效防止其在运输时激烈活动造成的伤害,从而增加运输距离和运输量,提高其存活率[3]。常用的渔用麻醉剂主要有MS-222、丁香酚类化合物、二氧化碳等[4]。其中,丁香酚以其高效、低价、代谢快的特点,受到广泛关注,但其对肝细胞具有一定毒性。目前,关于丁香酚对人体的危害及作为渔用麻醉剂的安全性在国际上仍具有一定的争议,日本、韩国、澳大利亚、新西兰、智利等国认定丁香酚是合法的渔用麻醉剂[5]。MS-222作为水生动物麻醉剂具有易处理、效力迅速、安全性高等特性[6-8],是美国FDA批准在指定种类水产品中使用的麻醉剂[9-11],但要经过21 d停药期才能上市销售。
目前, 中国对丁香酚类、MS-222等麻醉剂在水产品中的使用并未制定相关的政策法规和限量标准。因为监管制度的滞后,国内现缺乏对鱼用麻醉剂使用安全性的权威判定,如不及时加以引导和监管,可能会越来越多地引起公众对鱼用麻醉剂安全性的担忧和恐慌,同时也不利于渔业经济的健康发展。但现有方法仅针对单一种类麻醉剂,且方法检出线较高,基质干扰较严重,因此,需开发制定快速、高效且适合中国国情的检测方法。本研究中,利用三重四级杆气质联用技术,建立了能同时检测水产品中7种麻醉剂的前处理方法和高灵敏度、抗基质干扰、高准确性和高稳定性的三重四级杆气质联用质谱仪(GC-MS/MS)检测方法。
1 材料与方法
1.1 材料
试验仪器:气相色谱-质谱/质谱仪(Agilent公司)、氮吹仪(Organomation公司)、涡旋混合器(IKA公司)、超纯水器(Milliporeco公司)、离心机(湘仪公司)和电子天平(METTLER公司)。
试验试剂:丁香酚、甲基丁香酚、异丁香酚标准品(Dr.Ehrenstorfer公司),乙酸丁香酚酯、甲基异丁香酚、三卡因标准品(CNW公司),乙酸异丁香酚酯标准品(Chroma Dex公司),3-氨基苯甲酸甲酯内标标准品(TCI公司),丁子香酚-D3内标标准品(Accu Standard公司)、Silica 500 mg/6 cc、PRiME HLB 200 mg/6 cc、HLB 500 mg/6 cc、MCX 60 mg/3 cc固相萃取柱(Waters公司),Dispersive SPE(Agilent公司,15 mL),Cleanert MAS-Q(迪马公司);乙腈、正己烷、乙酸乙酯和丙酮为色谱纯,无水硫酸钠为分析纯(经550 ℃干燥4 h),所有用水均为超纯水。
1.2 方法
1.2.1 试验溶液的配制
标准储备溶液(1 mg/mL):准确称取丁香酚、甲基丁香酚、异丁香酚、三卡因、甲基异丁香酚、乙酸丁香酚酯、乙酸异丁香酚酯、丁香酚-D3、3-氨基苯甲酸甲酯7种标准品各10 mg,分别用乙腈溶解后定容至10 mL,配制成1 mg/mL标准储备溶液,置于冰箱(-18 ℃)中冷藏保存。
标准稀释溶液(10.0 μg/mL):准确吸取7种麻醉剂标准储备溶液各100 μL,用乙腈定容至10 mL,配制成10.0 μg/mL标准稀释溶液,置于冰箱(4 ℃)中冷藏保存。
内标稀释溶液(10.0 μg/mL):准确吸取两种麻醉剂内标储备溶液各100 μL,用乙腈定容至10 mL,配制成10.0 μg/mL内标稀释溶液,置于冰箱(4 ℃)中冷藏保存。
混合标准工作液:分别吸取适量的标准溶液和内标溶液,用乙酸乙酯与乙腈的混合液(体积比为3∶2)稀释,使标准品终浓度为1、5、10、25、50、75、100 μg/L,内标浓度均为10.0 μg/L,现用现配。
1.2.2 前处理方法 参照GB/T 30891—2014采样方法[12],称取5.00 g样品,置于50 mL离心管中,准确加入1 μg/mL的内标溶液50 μL,再依次加入5 g无水硫酸钠、10 mL正己烷及两粒陶瓷均质子,涡旋混匀1 min,超声提取15 min,以8000 r/min离心5 min,取上清液至干净离心管中,残渣用5 mL正己烷重复提取1次,合并上清液,待固相萃取净化。
将样品提取液全部加入硅胶柱(6 mL正己烷活化)中,控制流速小于0.5 mL/min,用2 mL正己烷淋洗,抽干后加8 mL乙酸乙酯洗脱,收集液体在低于45 ℃条件下,用氮气吹至3 mL,然后用乙腈定容至5 mL,涡旋混合1 min,过0.22 μm有机微孔滤膜,滤液供GC-MS/MS测定。
1.2.3 仪器条件
GC条件:载气为高纯氦气(99.9995%),恒流流速为1.5 mL/min;进样口温度为280 ℃,传输线温度为260 ℃;升温程序为初始温度为80 ℃, 保持0.5 min,然后以8 ℃/min升至152 ℃,保持0 min,再以2 ℃/min升至160 ℃, 并保持0 min, 最后以60 ℃/min升至270 ℃,保持4 min;进样方式为不分流,进样体积为1.0 μL;色谱柱为HP-5MS UI毛细管柱(30 m×0.320 mm×0.25 μm)。
MS-MS条件:离子源为EI源,离子源温度为230 ℃;离子化能量为70 eV;溶剂延迟为5.0 min;四级杆温度为150 ℃;淬灭气为高纯氦气,2.25 mL/min;碰撞气(用于MRM)为高纯氮气,1.5 mL/min。
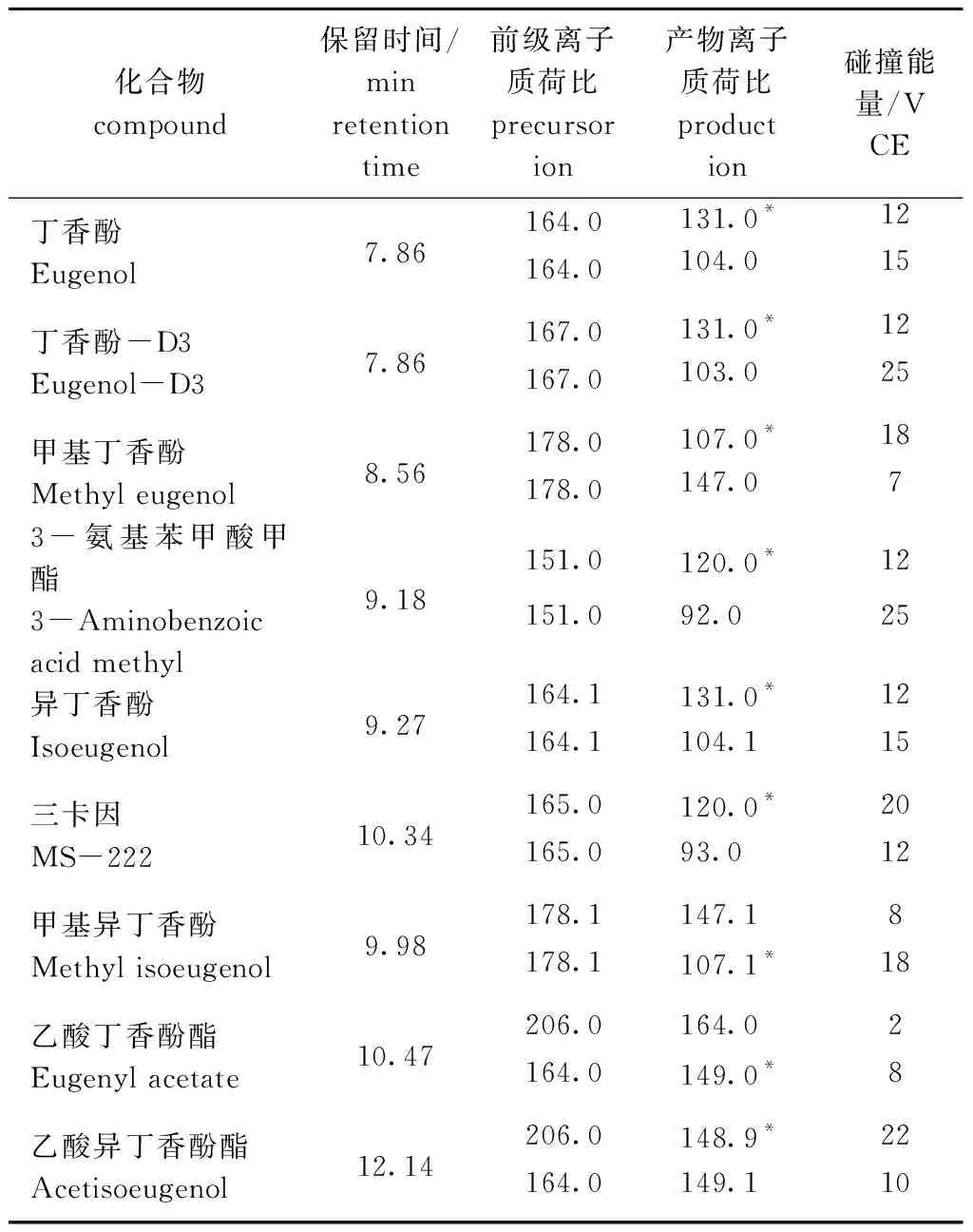
在选定的色谱条件下,通过分析7种麻醉剂标准溶液的单杆全扫描(MS1 Scan)质谱得到每种物质的总离子流图,选择质荷比(m/z)和相对丰度较高的一级碎片为目标化合物的母离子,确定保留时间和前级离子;在产物离子扫描(Product Ion)模式下,设定合适的碰撞电压,使目标物母离子与碰撞气体(高纯氮气)进行碰撞诱导解离,进一步裂解产生碎片离子得到二级质谱图,分别选取丰度较强且基质干扰较小的碎片离子作为定量和定性子离子;选择电子轰击离子源多反应监测模式(MRM),进一步优化碰撞能量,使特征碎片离子产生的离子对强度均达到最大,各目标物最佳参数见表1。
表17种麻醉剂及其内标的离子监测参数
Tab.1Monitoringparametersofionsandinternalcompoundsin7anesthetics
化合物compound保留时间/minretention time 前级离子质荷比precursor ion产物离子质荷比product ion碰撞能量/VCE丁香酚 Eugenol7.86164.0131.0*12164.0104.015丁香酚-D3 Eugenol-D37.86167.0131.0*12167.0103.025甲基丁香酚 Methyl eugenol8.56178.0107.0*18178.0147.073-氨基苯甲酸甲酯3-Aminobenzoic acid methyl9.18151.0120.0*12151.092.025异丁香酚 Isoeugenol9.27164.1131.0*12164.1104.115三卡因 MS-22210.34165.0120.0*20165.093.012甲基异丁香酚 Methyl isoeugenol9.98178.1147.18178.1107.1*18乙酸丁香酚酯 Eugenyl acetate10.47206.0164.02164.0149.0*8乙酸异丁香酚酯 Acetisoeugenol12.14206.0148.9*22164.0149.110
注:*为定量离子
Note: *denotes quantitative ions
2 结果与分析
2.1 总离子流图
在“1.2.3”节仪器条件下,鱼肉加标样品的总离子流图如图1所示,13 min内所有目标化合物均已出峰,目标化合物不仅达到完全分离,峰形良好,且在目标物出峰处无杂质干扰。因此,可选择三重四级杆气质联用法。

图1 MRM模式下鱼肉加标样品总离子流图Fig.1 TIC of derivatives in 7 anesthetic residues in fish by MRM mode
2.2 线性关系和检出限、定量限
在“1.2.3”节仪器条件下,对标准溶液进行进样分析,以目标物浓度(x)为横坐标,其峰面积(y)为纵坐标绘制标准曲线,计算回归方程和相关系数(表2),其相关系数R2均大于0.999,说明0~100 μg/L 范围内7种麻醉剂的浓度和峰面积线性关系良好。

表2 鱼肉中7种麻醉剂线性回归方程及其相关系数Tab.2 Linear equations and R2 in 7 anesthetics in fish
依据定性、定量离子色谱峰的信噪比(S/N)大于3为检出限(LOD),以S/N大于10为定量限(LOQ),确定本方法中7种麻醉剂的检出限为2.0 μg/kg,定量限为5.0 μg/kg。
2.3 准确度及精密度
在空白样品中添加待测物,使之理论含量为5.0、10.0、50.0 μg/kg,经上述试验步骤处理后,对3个水平进行回收率测试(表3),每个浓度平行测定6次,分别考察3 d,以相对标准偏差(RSD)计算批内和批间精密度。结果表明,此方法的回收率为73.93%~119.97%,且相对标准偏差满足GB/T 27404—2008[13]要求,说明其准确度和精密度较高,可以满足日常检测需求。
2.4 实际样品分析
用上述方法分别检测市售鲜活淡水鱼、虾类(包括黑鱼、鲫、草鱼、河虾等)和海水鱼、虾、贝、蟹类(包括鳕、黄鱼、多宝鱼、石斑鱼、斑节虾、花蛤、梭子蟹等)35个样品,考察试验结果,其中一批草鱼、两批鳕、一批长江回鱼、一批金鲳鱼和一批千岛湖鲫共6批次样品中检出丁香酚类麻醉剂,检出率为17.1%,最高残留量达298 μg/kg(长江回鱼);MS-222均未被检出。
3 讨论
3.1 净化方法的选择
丁香酚类微溶于水而易溶于有机溶剂[14],MS-222易溶于水[15],两种类型麻醉剂的理化特性完全不同,同时提取净化是个难点。本试验中考察了不同萃取方式,结果表明,采用HLB小柱净化后6种丁香酚类化合物均损失严重,MS-222的回收率偏低;6种丁香酚类化合物在MCX小柱的回收率优于HLB小柱,但结果偏低;采用PRiME HLB小柱和硅胶柱处理后,6种丁香酚类化合物回收率可以满足检测要求,但是MS-222经PRiME HLB小柱处理后损失较大;而采用硅胶柱对所有目标物都有较好的保留;此外,尝试Dispersive SPE和Cleanert MAS-Q两种不同分散固相萃取方法,回收率均可以满足要求,但是后期试验发现,分散固相萃取方法的净化效果没有硅胶柱好,因此,本试验的前处理采用硅胶柱进行富集净化。
3.2 色谱柱的选择
参照相关资料,并考虑方法的通用性,本试验中选用甲基聚硅氧烷固定相的DB-5MS毛细管色谱柱,7种麻醉剂在该色谱柱的分离效果如图2所示,丁香酚、异丁香酚和MS-222出现明显的拖尾,且化合物出峰时间集中在4 min内,分离效果不理想;而改用HP-5MS UI毛细管柱后,在相同仪器条件下,目标物的峰宽均小于0.12 min,拖尾现象得到改善,基线噪音显著降低,目标化合物峰形对称且其分离度和灵敏度均有所提高。故本试验中最终采用HP-5MS UI毛细管柱。
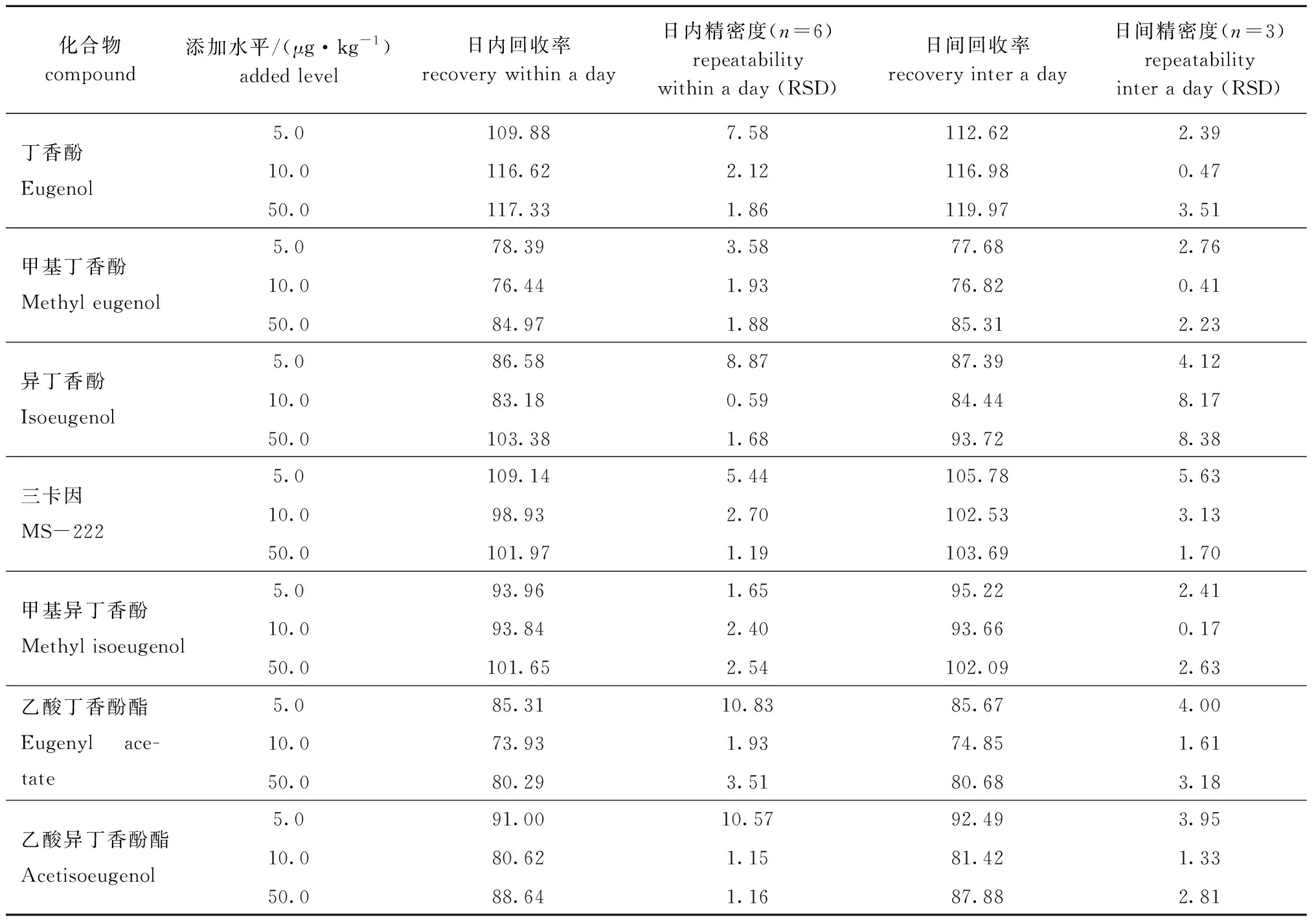
表3 鱼肉中7种麻醉剂的回收率和精密度(相对标准偏差)Tab.3 Recoveries and repeatability (RSD) of 7 anesthetics in fish %

图2 不同色谱柱中7种麻醉剂的色谱图Fig.2 Chromatograms of 7 anesthetic residues by different columns
3.3 仪器方法的选择
目前,国内已报道的检测方法中,仅检测单一种类的丁香酚类化合物[16],主要有液相色谱法[17-18]、液相色谱-质谱联用法[19]、气相色谱-质谱联用法[20-21]。对于MS-222残留检测,较常见的是液相色谱-质谱联用法[22-23]和气相色谱-质谱联用法[10]。本试验中采用三重四级杆气质联用质谱分析方法,7种目标物和两种内标在多反应监测(MRM)模式下的质谱图见图3。
本实验室前期试验比对了液相色谱-质谱联用法、气质联用法和三重四级杆气质联用法的检测效果。液相色谱-质谱联用法仅适用于MS-222残留检测,而丁香酚类由于挥发性太强、沸点低,在液质上碎片离子较小容易受干扰,检测结果不稳定。用气质联用法能同时分析7种麻醉剂,但检出限较高,基质干扰明显。丁香酚和其同位素内标丁香酚-D3打碎后得到了相同特征碎片离子,由于保留时间相同,在气质中无法区分,而在三重四级杆气质中,两物质的前级离子不同,即使其产物离子和保留时间均一致,也可以完全区分开;此外,质谱数据采用分段多反应监测模式,根据目标物出峰时间分成4段,缩短扫描所需的循环时间,使每个峰上得到的数据点数增多,大大提高了分析通量,确保了结果的稳定性和灵敏度,7种麻醉剂检出限均可达到2 μg/kg,而使用普通气质联用法时丁香酚检出限为5 μg/kg[24]。

◆表示前级离子;△▲表示产物离子Note:◆,precursor ion;△▲,product ion图3 MRM模式下7种麻醉剂及其内标的质谱图Fig.3 Mass spectrum of 7 anesthetic residues and internal compounds by MRM mode
4 结论
本试验中基于气相色谱联用三重四级杆质谱分析技术,建立的GC-MS/MS检测方法,采用多反应监测模式,根据保留时间、特征离子及其丰度对比,可对鱼虾等水产品中7种麻醉剂同时进行筛选、定量和确证,具有灵敏度高、抗基质干扰、高准确性和高稳定性的特点,能满足水产品中丁香酚类和MS-222麻醉剂残留量的检测要求,对保障消费者的健康,维护中国水产品的正常进出口贸易具有一定的意义。
