淫羊藿总黄酮提取物的HPLC指纹图谱建立及其中8种成分的含量测定Δ
2019-01-02牛晓静孙广科段晓颖徐立然
牛晓静,鲁 静,孙广科,段晓颖,徐立然
(河南中医药大学第一附属医院,郑州 450008)
组分中药是指由有效组分配伍而成的现代中药,是创新中药研究的一种途径,其特点是“两个相对清楚”,即物质基础相对清楚及其作用机制相对清楚;组分中药具有满足现代药物质量可控要求,安全性、有效性证据较充分的特征,既保持了中药方剂的优势,又提高了中药制剂的质控水平[1-4]。本课题组对益元康方按照组分中药的研究思路开展基础研究,就该方组成药材之一淫羊藿中主要有效成分黄酮类成分进行提取、分离、纯化,得到淫羊藿总黄酮提取物。本研究中,本课题组采用高效液相色谱法(HPLC)建立了淫羊藿总黄酮提取物的指纹图谱,并对其中8种成分的含量进行了测定,旨在为有效控制其质量提供参考。
1 材料
1.1 仪器
1260型HPLC仪,包括四元泵、二极管阵列检测器、在线脱气装置、自动进样器、Openl AB色谱工作站(美国安捷伦公司);CP225D型电子天平、BSA224S-CW型电子天平(德国赛多利斯公司)。
1.2 药品与试剂
淫羊藿总黄酮提取物(河南中医药大学第一附属医院中药制剂实验室自制,编号:G-1、G-2、G-3、G-4、G-5,总黄酮平均含量:71.6%);淫羊藿苷对照品(批号:110737-200415,供含量测定用)、宝藿苷Ⅰ对照品(批号:111852-201102,供含量测定用)均由中国食品药品检定研究院提供;淫羊藿属苷A对照品(批号:151023,纯度:≥98%)、朝藿定A1对照品(批号:16071302,纯度:≥98%)、朝藿定A对照品(批号:130520,纯度:≥98%)、朝藿定B对照品(批号:130518,纯度:≥98%)、朝藿定C对照品(批号:130601,纯度:≥98%)、鼠李糖基淫羊藿次苷Ⅱ对照品(批号:151023,纯度:≥98%)均由成都普菲德生物技术有限公司提供;甲醇、乙腈为色谱纯,其他试剂均为分析纯,水为纯化水。
2 方法与结果
2.1 色谱条件
色谱柱:ZORBAX Eclispse SB-C18(250 mm×4.6 mm,5µm);流动相:乙腈(A)-水(B)为流动相,梯度洗脱(0~8 min,15%A→20%A;8~25 min,20%A→23%A;25~40 min,23%A→30%A;40→50 min,30%A→32%A;50~68 min,32%A→35%A;68~75 min,35%A→45%A;75~80 min,45%A→60%A;80~85 min,60%A→15%A);流速:1.0 mL/min;柱温:30℃;检测波长:270 nm;进样量:5µL。
2.2 溶液的制备
2.2.1 单一对照品贮备液 精密称取淫羊藿属苷A、朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、鼠李糖基淫羊藿次苷Ⅱ、宝藿苷Ⅰ对照品各适量,分别置于25 mL量瓶中,加甲醇溶解并稀释制成质量浓度分别为0.390、0.282、1.010、1.338、1.792、1.600、0.268、0.908 mg/mL的单一对照品贮备液。
2.2.2 混合对照品溶液 分别精密量取“2.2.1”项下单一对照品贮备液各适量,置于同一10 mL量瓶中,加甲醇定容,摇匀,得含淫羊藿属苷A、朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、鼠李糖基淫羊藿次苷Ⅱ、宝藿苷Ⅰ质量浓度分别为 19.50、28.20、50.50、66.90、89.60、160.00、26.80、27.24 µg/mL的混合对照品溶液。
2.2.3 供试品溶液 取样品20 mg,精密称定,置于锥形瓶中,加甲醇20 mL振摇使溶解,经0.45 μm微孔滤膜滤过,取续滤液,即得。
2.3 方法学考察
2.3.1 精密度试验 取“2.2.3”项下供试品溶液(编号:G-1)适量,按“2.1”项下色谱条件连续进样测定6次,以淫羊藿苷峰的保留时间和峰面积为参照,记录各共有峰的相对保留时间和相对峰面积。结果,22个共有峰相对保留时间和相对峰面积的RSD均小于3%(n=6),表明本方法精密度良好。
2.3.2 稳定性试验 取“2.2.3”项下供试品溶液(编号:G-1)适量,分别于室温下放置0、1、2、4、8、12、24 h时按“2.1”项下色谱条件进样测定,以淫羊藿苷峰的保留时间和峰面积为参照,记录各共有峰的相对保留时间和相对峰面积。结果,22个共有峰相对保留时间和相对峰面积的RSD均小于4%(n=7),表明供试品溶液于室温下放置24 h内基本稳定。
2.3.3 重复性试验 精密称取样品(编号:G-1)适量,共6份,按“2.2.3”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,以淫羊藿苷峰的保留时间和峰面积为参照,记录各共有峰的相对保留时间和相对峰面积。结果,22个共有峰相对保留时间和相对峰面积的RSD均小于5%(n=6),表明本方法重复性良好。
2.4 HPLC指纹图谱的生成与相似度评价、共有峰相关分析
2.4.1 HPLC指纹图谱的生成 取5批样品各适量,按“2.2.3”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,采用《中药色谱指纹图谱相似度评价系统(2004 A版)》对5批样品的HPLC图谱进行分析,得HPLC指纹图谱,详见图1、图2。
2.4.2 相似度评价 采用《中药色谱指纹图谱相似度评价系统(2004 A版)》,以样品的HPLC对照指纹图谱为对照,进行整体相似度评价。结果显示,5批样品的相似度均大于0.99,表明各批样品间差异较小,质量稳定性良好,详见表1。
2.4.3 共有峰的指认及相关分析 5批样品有22个共有峰,其峰面积合计占色谱峰总面积的90%以上。通过与对照品HPLC图谱比对,指认了8个共有峰分别为淫羊藿属苷A、朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、鼠李糖基淫羊藿次苷Ⅱ、宝藿苷Ⅰ。其中,20号峰为淫羊藿苷峰,由于其分离度较好、响应值较高,故以其为参照峰,计算其他峰相对于淫羊藿苷峰的相对保留时间和相对峰面积,详见表2、表3。
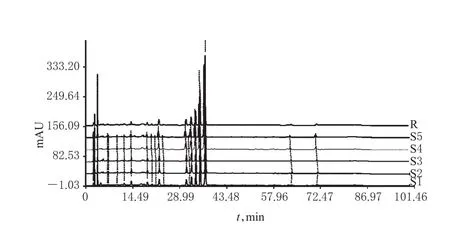
图1 5批样品的HPLC叠加指纹图谱Fig 1 HPLC superposed fingerprints of 5 batches of samples

图2 样品的HPLC对照指纹图谱Fig 2 HPLC control fingerprint of sample
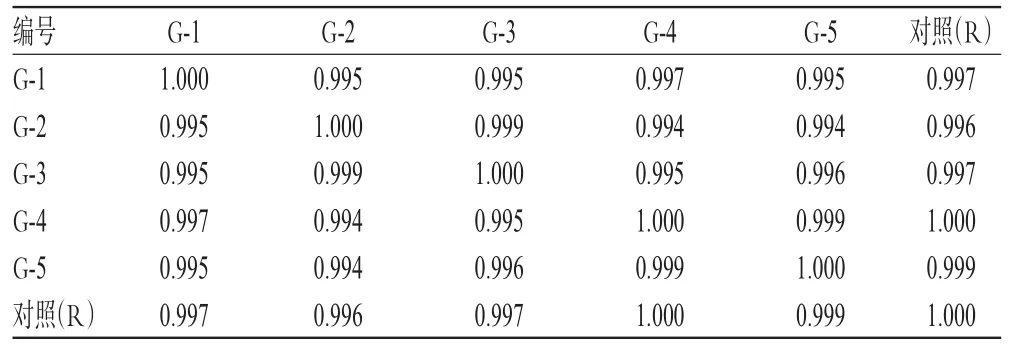
表1 5批样品相似度评价结果Tab 1 Results of similarity evaluation for 5 batches of samples
2.5 含量测定
2.5.1 系统适用性 取“2.2”项下混合对照品溶液、供试品溶液各适量,按“2.1”项下色谱条件进样测定,记录色谱图,详见图3。由图3可知,8种成分峰分离度均大于1.5,理论板数均大于5 000。
2.5.2 线性关系考察 分别精密吸取“2.2.2”项下混合对照品溶液1、2、5、10、15 μL,按“2.1”项下色谱条件进样测定,记录峰面积。以各待测成分进样量(x,μg)为横坐标、峰面积(y)为纵坐标进行线性回归,回归方程与线性范围见表4。
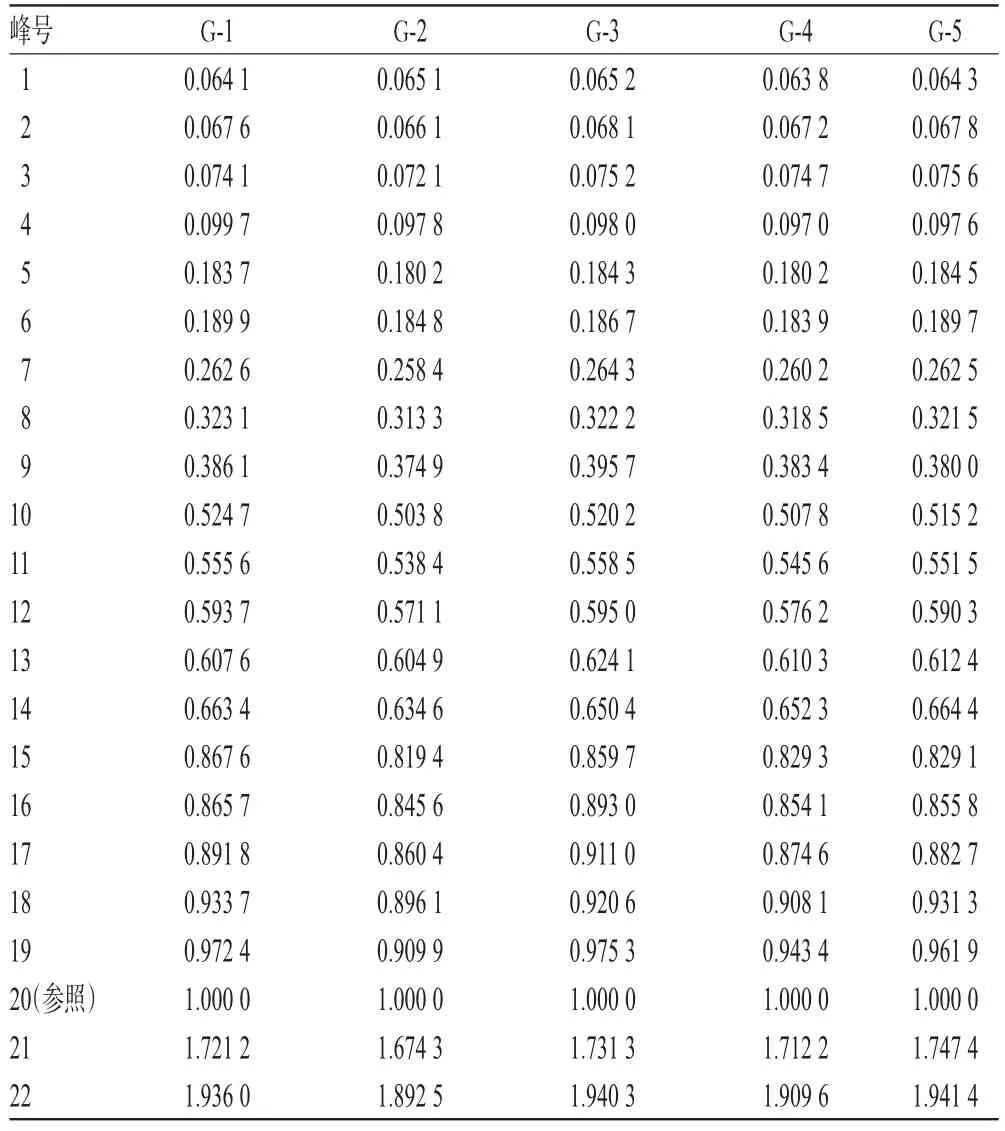
表2 5批样品HPLC图谱共有峰的相对保留时间Tab 2 Relative retention time of common peaks in HPLC chromatograms from 5 batches of samples

表3 5批样品HPLC图谱共有峰的相对峰面积Tab 3 Relative peak areas of common peaks in HPLC chromatograms from 5 batches of samples
2.5.3 定量限与检测限考察 分别精密吸取“2.2.2”项下混合对照品溶液适量,倍比稀释,按“2.1”项下色谱条件进样测定,记录峰面积,以信噪比10∶1、3∶1分别计算定量限、检测限。结果,淫羊藿属苷A、朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、鼠李糖基淫羊藿次苷Ⅱ、宝藿苷Ⅰ的定量限分别为390.00、564.00、506.00、535.20、448.00、426.68、643.20、544.80 ng/mL,检测限分别为97.50、141.00、126.25、133.80、112.00、106.67、160.80、136.20 ng/mL。
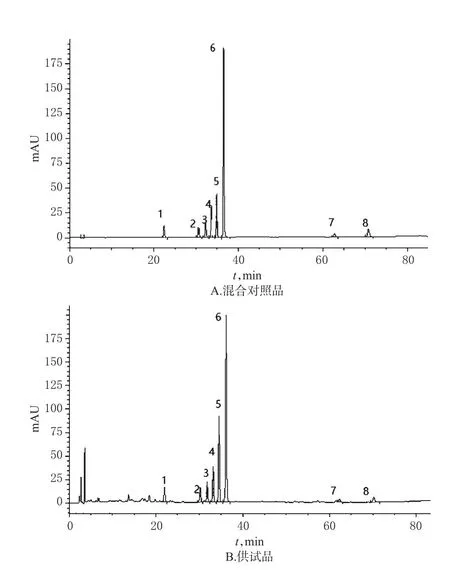
图3 高效液相色谱图Fig 3 HPLC chromatograms
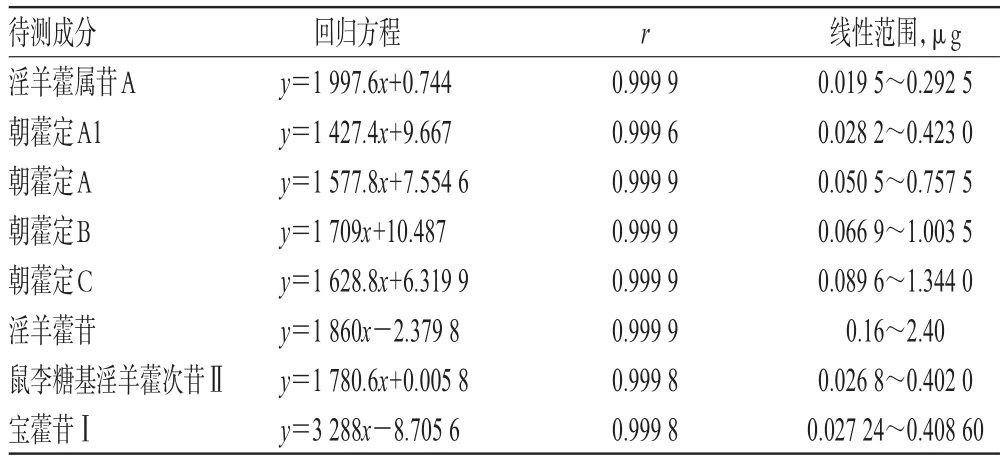
表4 回归方程与线性范围Tab 4 Regression equation and linear range
2.5.4 精密度试验 取“2.2.3”项下供试品溶液(编号:G-5)适量,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,淫羊藿属苷A、朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、鼠李糖基淫羊藿次苷Ⅱ、宝藿苷Ⅰ峰面积的RSD分别为1.15%、1.15%、0.15%、0.13%、0.08%、0.10%、0.33%、1.46%(n=6),表明本方法精密度良好。
2.5.5 稳定性试验 取“2.2.3”项下供试品溶液(编号:G-5)适量,分别于室温下放置0、2、4、6、8、10、12 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,淫羊藿属苷A、朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、鼠李糖基淫羊藿次苷Ⅱ、宝藿苷Ⅰ峰面积的RSD分别为1.31%、1.25%、0.15%、0.12%、0.08%、0.09%、0.31%、1.31%(n=7),表明供试品溶液于室温下放置12 h内基本稳定。
2.5.6 重复性试验 取样品(编号:G-5)适量,共6份,按“2.2.3”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积。结果,淫羊藿属苷A、朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、鼠李糖基淫羊藿次苷Ⅱ、宝藿苷Ⅰ的平均含量分别为2.94%、4.09%、4.76%、7.10%、17.33%、34.49%、0.89%、1.01%,RSD分别为2.04%、2.97%、2.93%、2.31%、2.16%、2.41%、1.61%、0.71%(n=6),表明本方法重复性良好。
2.5.7 加样回收率试验 取已知含量的样品(编号:G-5)10 mg,共6份,分别加入“2.2.1”项下单一对照品贮备液(淫羊藿属苷A、朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、鼠李糖基淫羊藿次苷Ⅱ、宝藿苷Ⅰ对照品贮备液分别加入750µL、1.5 mL、400µL、550µL、950µL、2 mL、330µL、110µL),按“2.2.3”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表5。

表5 加样回收率试验结果(n=6)Tab 5Results of recovery tests(n=6)
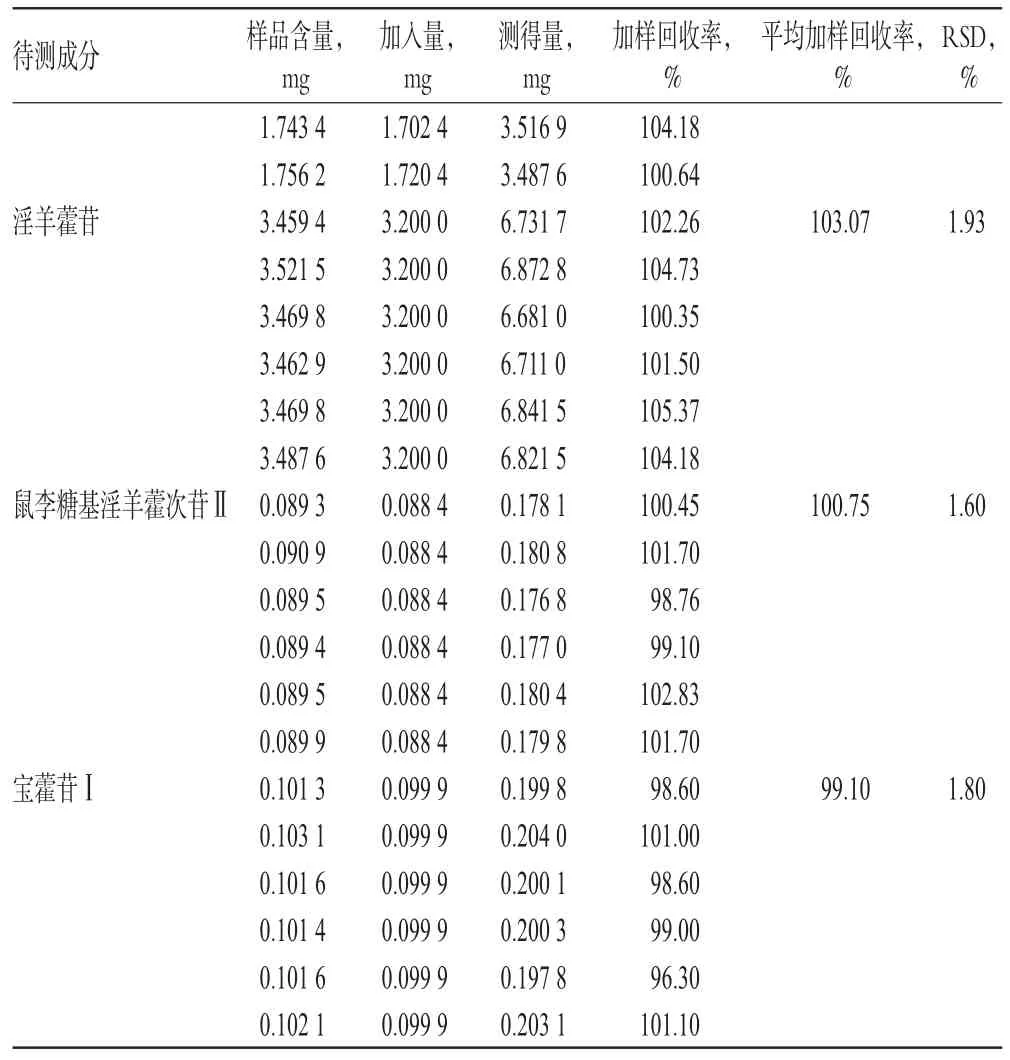
续表5Continued tab 5
2.5.8 样品含量测定 取5批样品各适量,分别按“2.2.3”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并以外标法计算其中8种成分的含量,结果见表6。
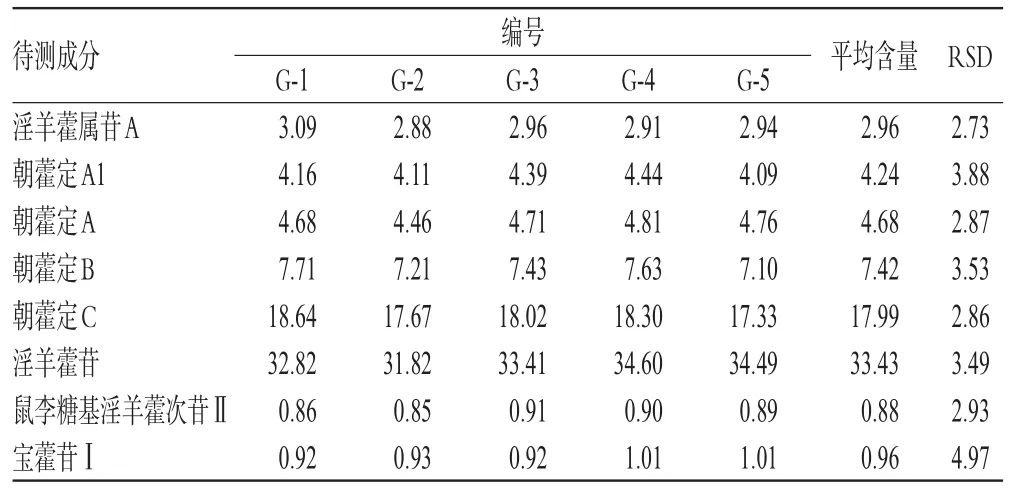
表6 样品含量测定结果(n=3,%%)Tab 6 Results of content determination of samples(n=3,%%)
3 讨论
本课题组前期通过均匀设计[5],发现以60%乙醇提取淫羊藿总黄酮的效果最佳,由于淫羊藿总黄酮提取物中存在大量杂质,故通过大孔吸附树脂HPD-400进行纯化除杂后,可获得纯度较高的总黄酮,经测定其总黄酮平均含量为71.6%。
本试验参考文献[6-10],采用HPLC法对5批已纯化的淫羊藿总黄酮提取物建立了指纹图谱,发现有22个共有峰,各批样品相似度均在0.99以上,质量稳定性良好。为进一步比较各批样品间的含量差异,选择标定指认的8个共有峰,建立含量测定方法。含量测定结果显示,5批样品中8种成分含量的RSD均小于5%,表明5批样品含量差异不大,提取及纯化工艺稳定、可行。
综上所述,所建HPLC指纹图谱可为淫羊藿总黄酮提取物的质量控制提供参考;所建含量测定方法简便、准确、重复性好,可用于同时测定淫羊藿总黄酮提取物中8种成分的含量。
