轨道序和巡游性对尖晶石结构钒氧化物AV2O4(A=Mn,Fe,Co)物性影响的研究
2018-12-28林高庭孙玉平
马 杰 ,林高庭 ,罗 轩 ,孙玉平
1上海交通大学物理与天文学院,上海,200240
2中国科学院固体物理研究所,合肥,230031
3中国科学技术大学,合肥,230026
4中国科学院强磁场科学中心,合肥,230031
5南京大学先进微结构协同创新中心,南京,210093
目录
I.引言239
II.实验方法 241
A.样品制备 241
B.测试技术 241
III.Mn1-xCoxV2O4242
A.磁化率和比热 242
B.X射线衍射 242
C.中子散射 242
IV.Fe1-xCoxV2O4247
A.磁化率和比热 247
B.X射线衍射 247
C.中子衍射 248
V.结论 250
致谢250
参考文献 250
I.引言
在强关联电子体系中,具有几何阻挫的 3d过渡金属氧化物不仅为凝聚态物理学理论研究的发展提供了良好的素材,而且由于其存在各种低温新奇的现象,引起了研究者广泛的兴趣[1-3]。 该体系通过调节轨道、自旋、晶格、电子等相关自由度之间的相互作用,会表现出一系列奇异的物理特性,如金属-绝缘体转变[4,5]、巨磁阻效应[6]、多铁效应[7-9]等。其中,因为库仑相互作用和泡利不相容原理而得到的磁相互作用的有效哈密顿量是由空间重叠的电子轨道决定的,所以电子的轨道自由度往往对低温奇异的物理特性起着重要的作用[10]。当轨道自由度出现简并,并与晶格自由度发生耦合[即我们通常所说的Jahn-Teller(J-T)畸变],轨道有序可以被诱发,从而对整个体系的磁性能产生很大的影响,比如在锰酸盐中出现的巨磁阻效应。因此,强的轨道关联对反铁磁绝缘体到铁磁金属的转变起了重要的作用[11]。如果自旋自由度同时也发生简并(即我们通常认为的磁阻挫现象),一系列复杂而有趣的物理现象就可以进一步的引发。
在这类体系的候选材料中,钙钛矿结构氧化物ABO3,尖晶石结构氧化物AB2O4以及烧绿石结构氧化物A2B2O7等等都是长久以来研究的热点[1-5],比如在钙钛矿锰氧化物中 Mn3+(3d4)的二重简并eg轨道[12],以及钙钛矿和尖晶石的钒基氧化物中 V3+(3d2)的三重简并t2g轨道[2,13,14]都被广泛研究。其中,尖晶石钒酸盐由于在没有轨道有序的情况下表现出完美的立方结构[2,14],所以占据了一个特殊的位置。与钙钛矿结构锰氧化物和钒氧化物所不同:在没有轨道有序时,钙钛矿锰氧化物的晶体结构仍可以发生一定的畸变(即 GdFeO3畸变)。而这种固有畸变的相应物理起源会使得轨道序的研究产生很大的干扰,因此,尖晶石钒基氧化物是一个研究轨道序起源的良好载体。
在正尖晶石AV2O4(A=Cd,Zn,Mg,Mn,Fe,Co)[2,14-32]中,B位离子的复杂结构往往使该体系表现出多种不同类型的相变,比如结构相变,磁相变,铁电有序等特征。如图1(a)所示,B位V3+离子可以单独构成共角四面体,即烧绿石结构,从而使得该体系有着较强的几何阻挫。另一方面,B位钒离子被6个氧包围形成 V-O八面体,所以 V3+的d轨道在八面体晶体场作用下发生劈裂,即五重简并的3d态分裂成为能量相对较低的三重简并态t2g和能量相对较高的二重简并态eg。这种共边V-O八面体可以导致最近邻V3+离子之间的轨道重叠,使得V3+之间的 Kugel-Khomskii型自旋轨道相互作用增强[33]。同时,轨道序也会被局域的V3+离子所诱发。然而,实验发现在 CoV2O4,ZnV2O4和 MgV2O4为代表的正尖晶石钒氧化物中,电子并不仅仅具有局域性,而是可以利用外加压力、掺杂等手段引入化学压力来影响其 V-V间距 (RV-V),从而实现对其导电性能的调控。也就是说,电子巡游性随着RV-V减小逐渐增强。理论研究进一步表明RV-V存在一个临界值:当Rc<2.94时,该体系将由半导体行为转变为金属行为[17,18,20-22,24,25,32,35-38]。

图1.(a)尖晶石AV2O4的晶体结构示意图;(b)随着温度变化FeV2O4的相演变规律。图中晶格参数随温度的变化关系源于参考文献[39,40]。需要注意的是高温四方相(HT Tetra)和低温四方相 (LT Tetra)晶轴的定义:HT=()(- ),HT=()(+ ), HT= , LT=(-)(+ ), LT=(-)(- ),LT= ,在这里 , , 代表晶格矢量,图中a,b和c对应了模的大小。
另外,处于 AO4四面体中心的 A位离子对AV2O4的物性也有一定的影响。简单来说,我们可以将AV2O4体系分成两组。一组以A位离子是非磁性的离子,即 A=Cd,Mg,Zn[16,26,28-31,35,41-43]。对于这三个材料,由于在低温时发生轨道有序,所以其相应晶体结构从立方相变为四方相;同时,V3+离子烧绿石结构造成的几何阻挫弱化,反铁磁序出现。另外一组是由磁性离子所组成,即A=Mn,Fe,Co,这样不仅引入了A-V之间的磁相互作用,而且可以引入J-T活性离子Fe2+(3d6),从而使得该体系的研究变得更为复杂。例如:(i)MnV2O4[44-51]在56 K发生共线亚铁磁(CL)相变,即Mn2+和V3+的磁矩出现共线反平行排列,但总的磁矩不为零。随后在53 K由于V3+在t2g轨道的有序产生反铁性轨道序,即dxy轨道被一个电子占据,而另外一个电子沿着c轴方向被dyz和dzx交替占据。早在 2003年,Tsunetsugu和 Motome提出了轨道有序与磁长程有序相关的模型,他们认为当 Kugel-Khomskii型自旋轨道相互作用增强时,反铁性轨道序将会出现[52]。并且,他们还认为该轨道序相变伴随着结构由立方相转变为四方相 (c<a),此外也导致 V3+的自旋沿着 [111]方向发生倾斜,即形成非共线亚铁磁序 (NC)。虽然他们以轨道有序来解释NC的成因看起来十分完美,但是这一模型在描述 CoV2O4时遇到了极大的挑战。(ii)CoV2O4分别在150 K和75 K发生了CL和NC相变,但是晶体结构始终保持着立方相,即没有结构相变发生[17-18,20,22,24,32,37-38]。目前认为,RV-V在CoV2O4中达到的最小值,更接近巡游电子极限,从而并没有发生轨道有序以及相应的结构相变。但是其NC的成因仍需要进一步理解。(iii)由于Fe2+离子在二重简并的eg态具有轨道自由度,这使得FeV2O4物性更丰富[19-20,25,39-40,53-56]。 其图解正如图 1(b)所示:首先,由于Fe2+离子在T1=140 K出现轨道序,从而导致了立方相向高温四方相(c<a)的晶体结构相变。这一结构相变来源于每个FeO4四面体之间的铁性轨道相互作用,导致集体 J-T效应。这个由于 3z2-r2(θ=180°)造成的轨道序有助于让一个压缩的四面体亚格子保持稳定,即形成高温四方相[19,39,40,56,57];然后,在T2=110 K发生从顺磁到CL的磁相变,同时伴随着四方相向正交相的结构相变,这个结构相变主要是由于自旋-轨道耦合所驱动的。由于在磁相变时导致体系出现强的自旋轨道耦合,从而当 Fe2+离子自旋有序排列时,会导致3d电子的x2-y2,y2-z2和z2-x2对应的轨道能量发生变化。根据 FeV2O4在 110 K下的磁结构,我们知道当Fe2+离子自旋沿着x轴排列时,将会有助于y2-z2(θ=120°)轨道趋于稳定,导致 3z2-r2和y2-z2的混合轨道态 (180°≥θ≥120°),因此,整个结构会出现沿着Fe2+离子自旋方向拉伸,即发生四方相向正交相的结构相变[39]。最后,在T3=60 K以下发生正交相向低温四方相(c>a)的结构相变,同时伴随着NC磁有序的出现,这个结构相变和NC都与另一个J-T活性离子V3+离子有着必然联系。在T3以下,V3+离子形成了一个复杂的轨道序[58]。在这里,由于沿着x轴压缩,导致t2g三重简并被分成低能yz单态和高能(xy+xz)的二重简并态,而这个二重简并态被V3+离子的另一个电子等价占据,即两个电子分别占据yz和轨道 (θ=300°)。而 FeV2O4出现低温四面体恰好有助于V3+离子的这个复杂的轨道序以及Fe2+的y2-z2轨道趋于稳定。此外由于自旋轨道耦合,V3+离子的自旋沿着<111>方向倾斜,从而形成NC磁结构[19,39]。这个独特的复杂铁性轨道序的形成很可能也是源于FeO4四面体和VO6八面体的角顶共享[39,59]:即在 FeO4和 VO6之间通过氧的连接使得轨道间相互作用有着反铁性的特征,从而引起FeO4四面体的拉伸进而导致临近VO6八面体的压缩,这种共同作用的结果会引起正交相向低温四方相的结构相变,造成V3+离子复杂轨道序的出现。
为了更好的理解在A位磁性离子的引入对AV2O4体系的结构和磁性的影响,我们对相应的Mn1-xCoxV2O4和 Fe1-xCoxV2O4体系进行了详细的研究。随着Co2+离子的掺杂,这两个体系均会接近巡游电子极限,从而表现出一些由于巡游性而造成的有趣的物性,比如轨道序的抑制,结构相变的消失,以及局域V3+离子引起的轨道序和巡游性造成的磁交换各向同性之间竞争的边界区的出现。我们的主要研究手段包括磁化率、比热、变温X射线衍射(XRD)和中子散射等相关实验技术手段,以及第一性原理计算的理论手段,以达到实验和理论的结合。本文主要通过以下几个部分来展开:在第二部分,介绍了样品制备和测试细节;第三部分将主要论述Mn1-xCoxV2O4体系的一些结果;第四部分将主要论述Fe1-xCoxV2O4体系的一些结果;第五部分将对结果进行总结。
II.实验方法
A.样品制备
Mn1-xCoxV2O4和Fe1-xCoxV2O4系列单晶通过光浮区法制备得到。晶体生长所需的籽晶和籽晶棒通过固相反应法得到。合适比例的MnO、CoO、FeO和V2O3被研磨在一起,在40 MPa的静水压下制成直径约6 mm,长度约60 mm的多晶棒,然后在真空密闭的石英管中,在950°C下,进行煅烧12 h。我们将多晶棒放入有氩气保护的红外加热图像光浮区炉中,并且里面装有两个卤素灯和双椭球镜。在生长过程中,其生长速度为15-30 mm/h,在25 rpm下以相反的方向旋转进料和籽晶棒。因为V2O3会在生长过程中蒸发,所以在原料配比阶段需要加入额外的V2O3。同时,调试合适的生长速度也是获得高质量单晶的关键。
B.测试技术
我们将小片的单晶样品研磨成粉末来进行变温XRD测试,用低温氦气压缩机组对其进行降温,最低温度可降至 10 K,并利用 FullProf软件对所测的XRD数据进行精修拟合。单晶的取向通过X射线Laue衍射进行定向。在超导量子干涉仪(SQUID)上,通过0.01 T的外加磁场进行DC磁化率测量。在多物性测量系统 (PPMS)上进行比热测量。磁化率和比热测量用的晶体并没有进行定向。我们在美国橡树岭国家实验室 (ORNL)的高通量同位素反应堆 (HFIR)和同步辐射中子源 (SNS),以及美国国家计量标准局的中子研究中心(NCNR),利用单晶衍射仪(HB-3A)、冷中子三轴光谱仪(CG-4C)和时间飞行谱仪(SEQUOIA和 DCS),沿着(H,H,L)平面进行中子衍射和非弹性中子散射实验。
III.Mn1-xCoxV2O4
A.磁化率和比热
图2给出了Mn1-xCoxV2O4体系的磁化率和比热随温度变化的关系。在x≤0.4时,比热数据存在两个明显的峰:(i)高温峰恰好对应于磁化率开始明显上升的位置,由此,我们可以很显然地判定在TC处发生了一个CL相变,即Mn(Co)的自旋应该是和V的自旋方向处于反平行排列状态;(ii)低温峰恰好对应于磁化率陡然下降的位置(即ZFC-零场降温后升温测试),并且该温度点TS从X射线衍射数据证实发生了结构相变。此外,磁化率曲线在ZFC和FC(加场降温后升温测试)之间的偏离现象表明可能出现自旋玻璃态的迹象。对于 0.5≤x≤0.7,比热上的高温峰依然存在,但是低温峰已经变得非常弱而且展宽很大。磁化率的ZFC在该温度点的下降趋势也变得放缓,不再那么陡峭。当0.8≤x≤1.0时,比热仅仅在TC附近有一个峰,低温峰完全消失,这一现象和下节的XRD测试结果基本一致。
B.X射线衍射
我们通过在T=280 K的粉末XRD测试,肯定了 Mn1-xCoxV2O4均表现为立方结构,其空间群为Fd¯3m。正如图 3(a)所示,其满足 V´egard定律,即晶格常数a随着x的增加呈线性变化。随着Co含量的增加(即化学压力的增加),我们也观察到计算得到的RV-V在降低。根据低温XRD实验,我们研究了该体系的结构变化。当0.1≤x≤0.7时,晶体结构由立方相转变为四方相,其对应的空间群为I41/amd,这和MnV2O4的结构类似。但是在高掺杂Co的实验中,即0.8≤x≤1.0,在低于10 K是也没有观察到结构相变,一直保持立方结构。

图 2.Mn1-xCoxV2O4体系随温度变化的磁化率和比热测试。图片来源于参考文献[37]。
我们将0.1≤x≤0.8区间的系列样品晶格常数随温度的变化绘制,如图4所示。在 0.1≤x≤0.7之间,我们可以观察到与比热和磁化率一致的结果,即随着Co含量的增加,TS向低温移动。在x=0.8处,10 K以上温度没有结构相变发生。为了便于体现化学压力效应,我们将x的变化通过1/RV-V替换得到在10 K下的图3(b)所示结果,即在四方相中,c/a比率随着Co含量的增加也增加。图4体现了晶格常数与掺杂和温度依赖关系。当x≥0.8,相应的从立方相到四方相的结构相变消失了。因此,我们认为随着Co含量的增加,晶体结构由长程的相变转变为相应的结构畸变,并且这种畸变随着Co的掺杂变得越来越弱。
C.中子散射
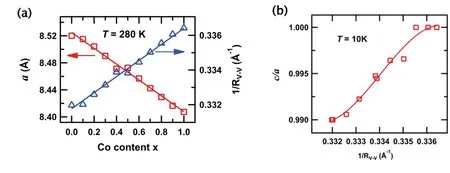
图3.(a)Mn1-xCoxV2O4体系在280 K下晶格常数a和1/RV-V随着x的变化;(b)在10 K下1/RV-V和c/a的比值的变化关系。图片来源于参考文献[37]。

图4.在 0.1≤x≤0.8系列样品中,晶格常数和温度的变化关系。图片来源于参考文献[37]。
单晶中子衍射实验测量了晶体结构、磁结构和轨道序与 Co掺杂的关系。图 5(a)和图 5(b)展示了 Mn1-xCoxV2O4的 (002)、(220)和 (004)的 Bragg峰与温度的关系。低于TC我们可以看到在对称性允许的Bragg位置(220)和(004)出现了亚铁磁信号。由于晶体对称性和散射强度的缘故,(002)衍射峰在四方相中不应该被观察到,而是仅仅存在于沿着ab面形成了反铁磁自旋NC结构(低于TNC)。因此,(002)是磁衍射峰,它的出现标志着在TNC处发生CL-NC的磁相变。另外,(004)衍射峰的强度也在TNC以下增加,这也提供了对相应磁相变的一个判据。当x=0.2时,由于在TC~70 K发生从顺磁向CL的磁相变,因此,(220)和 (004)的 Bragg峰的强度增强。在大约50 K(TS=TNC)附近,由于立方相到四方相的结构相变,(220)峰的强度明显下降。在x≤0.2时,TNC和TS是相等的。对于 0.2≤x≤0.8,TS<TNC。在x≥0.8,虽然XRD与比热测试均证实了晶体结构相变的消失,但是磁相变仍然存在,相应的温度分别为TC~150 K和TNC~80 K。
图 5(c)显示了 Mn1-xCoxV2O4随着 Co2+含量变化的相图:当x≤0.2时,在TC处发生顺磁到CL磁相变,在TS=TNC<TC处出现立方相向四方相的结构相变同时伴随着CL到NC的磁相变;当0.2<x<0.8时,CL,NC以及结构相变温度被分开,即TS<TNC<TC;当x≥0.8时,低至 5 K并没有出现结构相变,但是两个磁相变一直存在,且有TNC<TC。随着Co2+含量的增加,TC和TNC增加但TS逐渐降低。
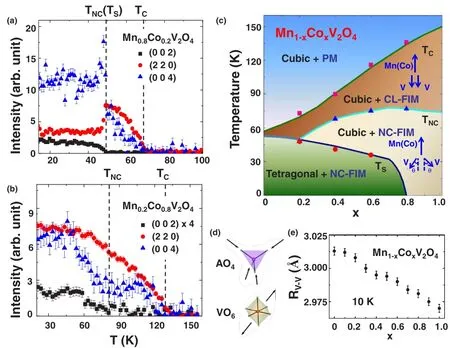
图5.在 Mn0.8Co0.2V2O4(a)和Mn0.2Co0.8V2O4(b)中Bragg峰(002)(方形)、(220)(圆形)和(004)(三角形)与温度的演变关系,在这里背景信号已经扣除;(c)温度和Co掺杂含量(x)的演变相图。通过磁化率或者比热测量(实线)以及中子散射实验(方形,三角形和圆形),我们得到对应的TC、TNC和TS三个相变温度;(d)AO4(A=Mn2+或者Co2+)四面体和VO6八面体的畸变发生方向;(e)在10 K下Co掺杂含量x与V-V间距RV-V之间的关系。图片来源于参考文献[22]。
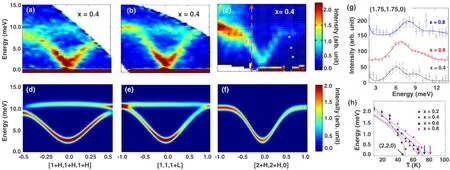
图 6.(a)-(c)在 8 K下 Mn0.6Co0.4V2O4的非弹性中子散射磁激发谱,在 (c)图中的箭头指的是截取的恒定 Q值的位置;(d)-(f)通过Eq.(1)计算得到的激发谱;(g)在8 K下,沿着[HH0],我们测得的在(1.75,1.75,0)下恒定Q值处不同样品的谱图特征。均采用Gauss拟合,随着Co的掺杂量的提高,低能自旋波分支发生硬化现象;(h)在磁区域中心(220)处的自旋波能隙。图片来源于参考文献[22]。
Co2+掺杂的微观机理可以定性的通过分析AO4四面体和 VO6八面体亚晶格的结构来理解。在高温立方相 (Fd¯3m)中,AO4四面体的内角 ∠O-A-O=109.7°。随着 Co2+的掺杂,A-O 键长从 2.041˚A(x=0)降低至 1.984˚A(x=0.8),沿着 A-O 键方向施加的化学压力正如图5(d)所示。此外,图5(d)也呈现了VO6八面体沿着<111>方向拉伸的示意图,这就造成了局域三角畸变,该畸变导致了12个O-VO内角从90°分成两类不等价的角。随着Co2+的掺杂,这两类不等价角之差从 12.3(2)°(x=0)降低至 10.0(2)°(x=0.8)。同时,V-O 键长从 2.023(1)˚A(x=0)缩短至2.012(1)˚A(x=0.8),V-V 键长 (RV-V)从 3.013 ˚A(x=0) 缩短至 2.975 ˚A(x=0.8),因此,化学压力和结构各向同性增加。
晶体结构相图反映了键长和键角随着Co含量变化的演变规律。当x≥0.8时,随着温度降低晶体结构的空间群从(立方)转变到I41/a(四方)。但是与很多尖晶石结构钒基氧化物都会发生结构相变[25,29,36,42,47,52,60]不同,当x≥0.8时,一直降至最低温(5 K)并没有观察到结构相变而是保持着Fd¯3m立方晶格。根据Tsunetsugu和Motome的理论,磁结构和晶体结构有着很强的关联性[52],因此,磁结构受到Co2+离子掺杂影响应该很大。低于TC高于TNC时,发生顺磁到反铁磁相变:i)Mn2+/Co2+离子磁矩的方向首先长程有序,并平行于c轴,同时,V3+离子的磁矩方向也是平行于c轴,但与Mn2+/Co2+离子的磁矩反向平行;ii)低于TNC时,V3+的磁矩类似于MnV2O4[50]和FeV2O4[10]的磁结构特征,沿c轴产生倾角。如果从V3+四面体的角度看,基态磁结构形成了二进/二出的自旋冰结构。具体表现为:V3+离子的磁矩低于70 K开始偏离c轴形成倾斜的结构,在 10 K下x=0.8时与c轴的夹角为 22.1±1.8°,小于该温度下的 33.7±1.5°(x=0)和 36.2±1.5°(x=0.2)。同时在 10 K下 V3+离子的磁矩从 0.95±0.04µB(x=0)增加到 1.03±0.07µB(x=0.2),然后降低至 0.61±0.03µB(x=0.8)。 在x≤0.2,随着 Co2+离子含量的增加 V3+离子磁矩升高,表明 Co2+离子的掺杂对 V3+离子的局域性有很大影响。正如图 5(c)所示,我们发现 V3+离子的磁矩在Co2+离子高掺杂区域(x≥0.8)并不会消失,并且其相应的磁相变温度随着掺杂量的增加而升高,表明之前的有关Co2+离子半径小从而导致顺磁性的理论模型过于简单,需要进一步的修正调整[36]。
为了确定NC态的起源是由巡游电子诱导的,我们利用非弹性中子散射谱 (INS)对自旋波进行了测量,并根据Heisenberg模型对磁性离子间相互交换作用和各向异性进行了评估。尽管在A2+离子之间的相互作用比较小[10,50,61],但是A2+离子和V3+离子之间的相互作用可以通过低能自旋波的色散来测定。正如图 6(a)-(c)所示,我们对x=0.4在 [111]布里渊区内,沿<111>、<001>、<110>等高对称方向的色散关系进行了测试。注意到图6(c)中两个磁模型被观察到,这与MnV2O4[10]的相同,并且自旋波速度随着Co2+离子的掺杂在增加。图6(h)展示了不同掺杂下在(220)区域中心的自旋波间隙能和温度的关系。正如MnV2O4[50]和FeV2O4[10]描述的那样,低于TNC自旋波的能隙是通过沿着每个V四面体的对角线产生的,这也使得V3+离子磁矩偏离了c轴。同时,在图 6(h)可以看到布里渊区中心的自旋波能隙在最低温(8K)时,几乎与Co2+离子的掺杂量无关。
在没有轨道序的情况下,自旋波理论可以用于理解巡游性驱动NC态的微观起源。这些计算是以六个不等价子格的哈密顿量为基础的,

不等价的 A位用下标p和q表示,不等价的 B位用下标i、j、k和l表示。最近邻的各向同性交换常数展示在图 7(c)中。A位自旋的易轴各向异性DA沿着c轴,然而B位的易轴各向异性DB沿着局域的<111>方向对应的值的范围产生了相似的匹配值。Mn0.6Co0.4Cr2O4所测得的实验数据是和由此得到最好的拟合参数值为
需要指出的是,根据 Heisenberg模型,布里渊区中心的自旋波能隙与成正比。因此,我们可以推断出与V3+离子的巡游性相关联的增强的JAB和被抑制的DB之间的平衡,导致了这一大约2 meV能隙。另外,被抑制的DB使得尖晶石结构的阻挫增强[62]。
正如图 6(d)-6(f)所示,Mn0.6Co0.4Cr2O4计算的色散关系与测试的数据吻合的比较好。相较于 MnV2O4的JAB来说,Mn0.6Co0.4Cr2O4的要更强。另外,密度泛函理论(DFT)表明通过引入电子巡游性,Co2+离子的掺杂也会加强晶体结构(晶格常数a≈b)和磁相互作用的各向同性,正如图7(a)和(b)所描述的那样。如果Co2+离子掺杂不增强JAB相互作用,V四面体对角线上剩余的磁各向异性可以将V自旋态转变为全进/全出结构。但是,由于JAB变强,相对于半径小的Co2+离子的掺杂,V自旋的基态仍然保持着之前的对称二进/二出态。

图 7.(a)和 (b)指的是 MnV2O4和 CoV2O4的电子态密度;(c)在 Mn1-xCoxV2O4体系中,非共线亚铁磁磁结构示意图。JAB,JabBB和JcBB最近邻位置之间的交换相互作用;(d)和 (e)指的是通过 DFT计算得到的轨道能量值,JA-V与能隙△呈反比关系,为了简化,仅展示了V3+离子的上自旋能级。图片来源于参考文献[22]。
如果用 DFT对A和B离子的轨道能量进行评估,可以解释JAB增强的起因,图解可参考图 7(d)和(e)。V3+离子和Mn2+离子在d轨道上耦合所需的大的能量差(~5eV)导致了V3+离子和Mn2+离子之间的交换作用被弱化。而Co2+离子的掺杂使得eg能级被填满,从而降低了t2g能级,最后导致V3+离子和Co2+离子的交换作用得到增强。DFT计算表明相比较MnV2O4(-1.2 meV)来说,反铁磁交换作用JAB在 CoV2O4(-2.5 meV)中明显增强。虽然 Co2+离子掺杂使 V3+离子的电子更具有巡游性,但增强的JAB有助于产生TC相变。此外,由轨道淬灭驱动(图7(d))的各向同性磁交换作用的增强能够稳定等对称的NC相,同时提高TNC相变温度。因此,即使没有轨道序的情况下,电子巡游性仍有助于CL和NC相的出现。
另外,需要指出的是诱导巡游性与RV-V是密切相关的。在10 K下,随着x增加至0.2,RV-V几乎保持不变;但是随着x进一步增大,RV-V开始下降。基于DFT计算我们发现更小的RV-V能够诱导电子的巡游行为(图7(a)和(b)),从而抑制轨道序。因为巡游性的出现可以进一步导致轨道序的消失,所以随着Co2+离子的掺杂导致TS相变温度下降。
如图8总结了不同x含量的晶体结构和自旋态的起源。对于低的Co2+离子掺杂,V3+离子的轨道序可以通过触发四方结构转变来缓解磁阻挫,并诱导出二进/二出的自旋基态。此时,JAB为Mn-V相互作用,只是增加了倾斜的角度但依然维持着二进/二出的自旋态。如果只考虑各向同性的V-V相互作用(JBB)和剩余的局域V3+离子各向异性,Co2+离子掺杂就会产生一种全进/全出的自旋态。但是,Co2+离子和V3+离子之间强的反铁磁交换作用JAB稳定了高Co2+离子掺杂化合物中观察到的二进二出的自旋态。因此,一些外界扰动(比如压力、磁场)会进一步加强电子的巡游性,削弱残余各向异性,增强磁阻挫。
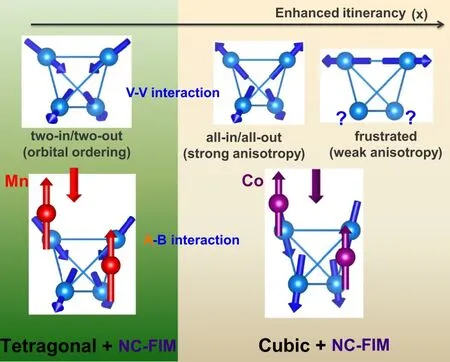
图8.在 Mn1-xCoxV2O4体系中,磁结构随着 x变化的演变示意图。图片来源于参考文献[22]。
IV.Fe1-xCoxV2O4
A.磁化率和比热
图9给出了Fe1-xCoxV2O4体系的磁化率和比热随温度变化的关系。对于x=0.05,比热上观察到三个明显的异常峰,对应的温度为T1=129 K,T2=108 K,T3=57 K,而磁化率在T2温度点处观察到明显增加的趋势,在T3处ZFC也有一个陡然的下降。该结果与FeV2O4(T1=139 K,T2=109 K,T3=60 K)的进行比照,可以知道在T1处发生了由立方相向高温四方相(c>a)的结构相变,5%的Co2+离子掺杂即可导致T1和T3向低温移动而T2却向高温移动的现象。对于x=0.1,比热上依然出现三个异常峰,磁化率ZFC曲线尽管在T2处依然明显上升,但是在T3处却表现出缓慢下降的趋势。在x=0.2时,比热上仅出现两个异常峰,因为在磁化率上T2温度点出现明显的上升,这恰好对应比热上的高温异常的温度点,因此我们将比热上的两个异常峰命名为T2和T3。在0.3≤x≤0.8,比热上仅出现T2这一个峰,很显然这依然是和磁化率明显上升相关的一个转变。而在x=0.9时,除了T2处发生异常之外,磁化率在T4=75 K处有一个明显的尖嘴状的异常。通过与CoV2O4进行比照,我们可以推断T2和T4的温度异常分别对应于CL和NC亚铁磁序。此外,为了更加突出磁化率ZFC曲线在0.1≤x≤0.7下T2和T3之间缓慢变化的一些细节以及一些可能忽视的异常,我们对ZFC曲线进行的一阶导数,正如图10所示。在 0.1≤x≤0.7,均在T5=60 K附近观察到一个比较展宽的异常峰。对于x=0.8,我们认为同时存在T4和T5这两个异常温度。
根据Fe1-xCoxV2O4体系比热和磁化率数据,我们可以观察到其伴随着复杂的磁序和结构的演变。随着Co2+离子的掺杂,我们可以得到以下变化:(i)T1先向低温移动,在x≥0.2之后消失;(ii)随着x的增加T2向高温移动;(iii)T3先向低温移动,在x≥0.3之后消失;(iv)T5(~60 K)在0.1≤x≤0.7貌似和Co2+离子的掺杂无关。
B.X射线衍射

图 9.Fe1-xCoxV2O4体系随温度变化的磁化率和比热测试。图片来源于参考文献[20]。

图10.Fe1-xCoxV2O4体系磁化率ZFC曲线的一阶导数。图片来源于参考文献[20]。
为了更好的理解Fe1-xCoxV2O4体系,结构随着温度的降低所表现的差异,我们研究了该体系的变温 XRD。 我们以x=0.1和x=0.5作为代表详细介绍一下这两个样品的结构演变过程,如图11所示。x=0.1时,随着温度降低至大约T1温度点附近,其结构从立方相转变为四方相(c>a)。温度进一步降低至T3时,出现晶格畸变,即晶格常数c和a分别发生了轻微的下降和增大,c/a的比率下降。有趣的是x=0.1的结果明显不同于x<0.1的结果,即 Fe1.1V1.9O4低于T1之后一直保持低温四方相 (c>a),并没有出现高温四方相(c<a)和正交结构相变,这些结果和x=0.2的类似。正如图11(b)所示,我们也对x=0.5的样品进行了详细研究。与x=0.1所不同的是结构相变温度T6(立方相向低温四方相 (c>a)转变)低于共线亚铁磁相变温度(T2),但是却高于T5,这些结果和0.3≤x≤0.6的类似。当x≥0.7,低至10 K,并没有观察到结构相变,而是一直保持立方结构。

图 11.(a)和 (b)分别表示 x=0.1和 x=0.5样品的晶格常数和 c/a比率随温度的演变关系。图片来源于参考文献[20]。
我们也对该体系在室温进行了XRD测试并进行精修得到图12所示结果,明显可以看出随着Co2+离子含量的增加,晶格常数a和RV-V呈降低趋势。而在10 K下,随着Co2+离子含量的增加,c/a的比率下降,表明了Co2+离子的掺杂使得该体系晶格畸变减弱。

图12.(a)室温下的晶格常数 a和dV-V以及 (b)10 K下的c/a随着Co掺杂的演变关系。图片来源于参考文献[20]。
C.中子衍射
我们也对Fe1-xCoxV2O4体系进行了单晶中子衍射测试,以便更加清晰的了解磁结构的演变。正如图13(a)所示,我们绘制了所选的这几个代表性样品的 (002),(220),(111)的 Bragg峰随温度的变化。相比较于FeV2O4,随着Co2+离子含量的增加,磁矩和 V3+离子的自旋倾斜角度均降低。然而x=0.2和0.5的样品,由于要经历一个结构相变,这导致很难确定其精确的磁矩和倾角大小。在5 K下x=0.8时,A位离子的总磁矩为 3.2(1)µB并且 V的倾角从 55(4)°(FeV2O4)下降到 38(3)°[39]。 对于x=0.2,当低于 109 K,在对称性允许的 (220)和 (111)开始上升,这说明出现亚铁磁信号,即发生了顺磁向共线亚铁磁的转变。在低于 60 K时,被对称性禁止的(002)峰出现,所以在ab面内却出现了反铁磁信号,即T5=60 K处发生了共线向非共线亚铁磁的转变。x=0.5和 0.8也在T5处出现 (002)。上述这些类似的现象也在x=0.5,0.8和0.9中观察到,因此,在T2处发生CL相变。需要强调的是在x=0.1和0.2时,T3不再代表CL向NC的相变,而是低温四方相的畸变;x=0.9时,发生CL向NC的温度是T4=75 K。
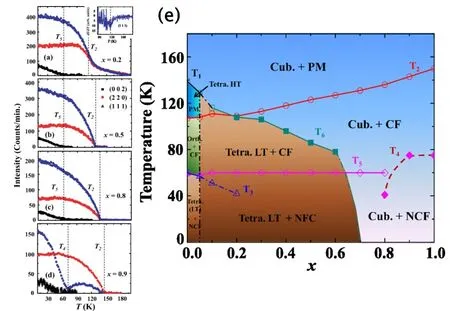
图 13.(a)-(d)表示在 HB-3A下测试了 Bragg峰 (002)(方形)、(220)(圆形)和 (111)(三角形)随温度的演变关系,(a)中的插图表示(111)Bragg峰的一阶导数;(e)温度和Co掺杂含量(x)的演变相图。T1()代表立方相向高温四方相(x=0.0和0.05)以及立方相向低温四方相(x=0.1)的相变;T2()代表顺磁向共线亚铁磁相变,同时也表示高温四方相向正交相(x=0.0和0.05)的相变;T3()代表正交相向低温四方相(x=0.0和0.05)的相变以及在低温四方相发生的晶格畸变 (x=0.1和 0.2);T4()代表 CL到 NC 的相变 (x≥0.8);T5()代表 CL到 NC 的相变 (x≤0.8);T6()代表立方相向低温四方相(0.2≤x≤0.6)的相变。图片来源于参考文献[20]。
正如展示在图13(b)中,从上述的一些测试结果我们可以归纳如下。首先,结构上在T1处的立方相向高温四方相(c<a)转变和在T2处的高温四方相向正交相的转变只出现在x=0和0.05;当x≥0.1时,T1、T2均消失。由此我们可以肯定T1和T2的结构相变主要受A位Fe2+离子来调控,且特别容易受到Co2+离子掺杂的影响。T1相变是由于J-T效应引起的FeO4四面体的压缩,而T2是由于Fe2+离子在磁有序处强的自旋轨道耦合造成的[39,40]。其次,x≥0.1,随着Co2+离子掺杂量的增加,顺磁向CL转变温度T2也增加。这种行为和 Mn1-xCoxV2O4体系的压力效应较为类似。我们知道随着 V-V间距的下降,Mn1-xCoxV2O4体系更接近巡游电子行为[37]。并且DFT对CoV2O4的研究表明,电子巡游性的增加导致磁各向异性减弱,同时A-B位之间的磁交换相互作用增强,因此有助于提高 CL转变温度[22]。再次,在x=0.1和0.05中,在稍微高于T2或者在其附近温度出现一个立方相向低温四方相(c>a)的结构相变,之后这个结构相变温度在0.2<x<0.7之间低于T2,我们将其命名为T6。当x≥0.7时,并没有再观察到结构相变。对于0.1≤x≤0.6,Co2+离子的掺杂造成的低温四方相的出现,可以认为主要是由V3+离子的轨道序诱发的。T2和T6的分离表明V3+离子的巡游性和轨道序之间的竞争。随着Co2+离子含量的增加,电子巡游性表现愈加明显,这使得磁有序温度向高温移动,而轨道序相变温度向低温移动,直至x≥0.7,轨道序被完全抑制。
在相图中我们也要注意到:(i)在0.0≤x≤0.8之间CL-NC磁相变温度T5=60 K和掺杂并无关系,而在x=0.9和 1.0下却跳到T4=75 K。对于x=0.0和 0.05,正交相向低温四方相的结构相变温度T3几乎和T5出现在统一温度,但是在 0.1≤x≤0.7之间并没有在T5处发生结构相变。这说明T5的磁相变主要受V3+来调控。而x=0.9和1.0时,主要由于更强的电子巡游性以及磁交换各向同性使得其在更高的温度T4=75 K处出现 CL-NC的转变[22]。从相图可以看出x=0.8恰好落在局域的 V3+离子造成的轨道序与巡游性造成的交换各向同性竞争的边界区。前者使得在T5处的NC趋于稳定,而后者使得在40 K处的NC趋于稳定,进而随着更高的Co2+离子掺杂使得T4升高。(ii)对于x=0.1和 0.2,并没有在T3处观察到结构相变,仅仅出现了微弱的晶格畸变造成c/a有所下降,,并且其出现在T5相变以下温度。在Fe1-xMnxV2O4体系也出现类似的特征,并认为该体系的这种晶格畸变是由于V3+离子的自旋倾斜过程所造成的自旋晶格耦合。而在我们的研究体系中,x=0.1和0.2在T3处的异常或许也是这种类似的原因。
V.结论
在此,我们对比了 Mn1-xCoxV2O4[22,24,37]和 Fe1-xCoxV2O4[20]以及 Fe1-xMnxV2O4[54]体系的异同点。在Fe1-xCoxV2O4和Fe1-xMnxV2O4的相似点是随着少量Co/Mn的掺杂,均会导致高温四方相(c<a)和正交相快速消失,相似的特征再次肯定了这两个结构相变完全受Fe2+离子的影响才出现的。这两者主要的区别是:(i)在Fe1-xMnxV2O4体系,在x≤0.6,顺磁到CL的相变总是伴随着立方相向低对称相(正交或者四方)的结构相变,并且在x≥0.7,CL向NC的磁相变总是伴随着正交相向另外一种四方相的结构相变。换句话说,就是在 Fe1-xMnxV2O4体系中自旋有序和结构相变有着很强的关联;(ii)有趣的是,在 Fe1-xCoxV2O4体系,Co2+离子的微量掺杂将导致磁相变温度和结构相变温度发生分离,即顺磁向 CL的磁相变温度在结构相变温度之上。同样,在 Mn1-xCoxV2O4体系中,随着 Co2+离子的微量掺杂也出现结构相变和磁相变的分离。即在母体MnV2O4中CL-NC的磁相变和结构相变在同一温度,随着 Co2+离子含量的增加,CL-NC的磁相变在结构相变温度之上。因此,我们可以定性的得出:在Co2+离子掺杂的体系总是会出现结构相变和磁相变的分离。这个分离应该主要归因于轨道序和巡游电子之间诱发的竞争机制导致的。随着Co2+离子含量的增加,电子巡游性的增强有助于A-B之间的磁交换相互作用和磁交换各向同性增强,这将导致磁相变温度向更高温度移动,同时使得轨道序弱化从而抑制晶格畸变,降低了结构相变温度。
总之,随着 Co2+离子含量的增加,Mn1-xCoxV2O4和 Fe1-xCoxV2O4体系将从局域电子行为转变为巡游电子行为,从而表现出丰富的物性特征。在低浓度的Co2+离子掺杂和高浓度的Co2+离子掺杂,其晶体结构和磁结构的演变表现出不同的物理起源。在Co2+离子低掺杂下,由于局域电子V3+离子产生的轨道序以及A位磁性离子的作用,往往在发生磁相变时伴随着结构相变的出现,甚至在A位出现J-T活性离子会导致顺磁区出现结构相变。但是随着Co2+离子的进一步掺杂,RV-V变短,引入电子巡游性的特征。这将弱化V3+离子形成的烧绿石亚格子的几何阻挫特性,从而弱化磁和结构各向异性,表现出明显的各向同性特征,同时诱发了局域电子轨道序和巡游电子之间的竞争。而巡游性的增强将逐步减弱轨道序的作用,直至整个体系不再发生结构相变。Co2+离子的引入也有助于JAB磁交换相互作用的增强,因此顺磁向亚铁磁的相变温度反而会向高温移动。通过我们对 Mn1-xCoxV2O4和 Fe1-xCoxV2O4体系的研究,将有助于对各种自由度之间相互竞争的微观图像有更加清晰的认识,这也为我们探索多种序参量同时出现提供了有效的研究思路。
致谢
国家自然科学基金/大科学装置联合基金培育项目,U1732154和 U1432139。
