在线固相萃取-超高效液相色谱/串联质谱法测定饮用水中8种精神类药物
2018-12-25,,,,,
, , , , ,
(北京出入境检验检疫局检验检疫技术中心,北京 100026)
目前,医药行业快速发展,据统计全球目前有4500多种药物被用于人类和动物的疾病预防和治疗[1]。这些药物尽管没有传统持久性有机污染物(POPs)的半衰期长,但随着人类的使用被持续不断的进入到环境中,使其在低浓度下产生的细微作用累积放大,对微生物、动植物和人类的生态毒性逐渐显现出来[2 - 3]。近些年,在地表水[4]、地下水[5]和饮用水[6]等多种水环境中检测出了多种精神类药物残留。Lindberg等[7]研究表明,污水处理厂对污水中的精神类药物降解率低,未去除的这类化合物经过地表与地下水循环体系污染人类的饮用水水源,给人类健康和生态环境造成严重的危害[8 - 9]。
近几年,为使国内抑郁症和癫痫患者得到有效治疗,这两类药物的生产和销量不断增加,因此,水环境中精神类药物污染情况开始受到人们的关注。但水中这类药物浓度较低,需要预富集以满足仪器分析要求。目前,水中抗抑郁药主要前处理富集方法包括液-液萃取法[10]、固相萃取法[11 - 12]和液-液微萃取法[13]。仪器检测方法主要包括液相色谱法[14]和液相色谱/串联质谱法[15]。本文采用在线固相萃取-超高效液相色谱/串联质谱法(OnlineSPE-UPLC-MS/MS)测定饮用水中常见的6种抗抑郁类药物阿米替林、舍曲林、帕罗西汀、阿利马嗪、氯米帕明和氟西汀和2种抗癫痫类(奥卡西平、卡马西平)。该方法快速准确,灵敏度高,并应用该方法对北京市部分饮用水源地采集的水样进行了检测,为饮用水的水质安全风险评估和法规制定提供必要的技术支撑。
1 实验部分
1.1 仪器与试剂
WatersAcquityXevoTQ-S液相色谱-串联四极杆质谱联用仪(美国,Waters公司);在线固相萃取装置(美国,Waters公司),配2777C自动进样器;尼龙材质针筒式微孔滤膜(直径13mm、0.22μm)(美国,PALL公司)。
甲醇、乙腈(色谱纯)(美国Fisher公司);甲酸(色谱纯)(美国Tedia公司)。实验用水为经Milli-Q净化系统(美国,Millipore公司)过滤的超纯水(18.2MΩ·cm)。
8种目标分析物标样信息详见表1(纯度≥95.0%,美国,Sigma-Aldrich),以甲醇溶液分别配制质量浓度为10mg/L的标准储备液,于-18℃保存,临用时配制成不同浓度的标准工作液。
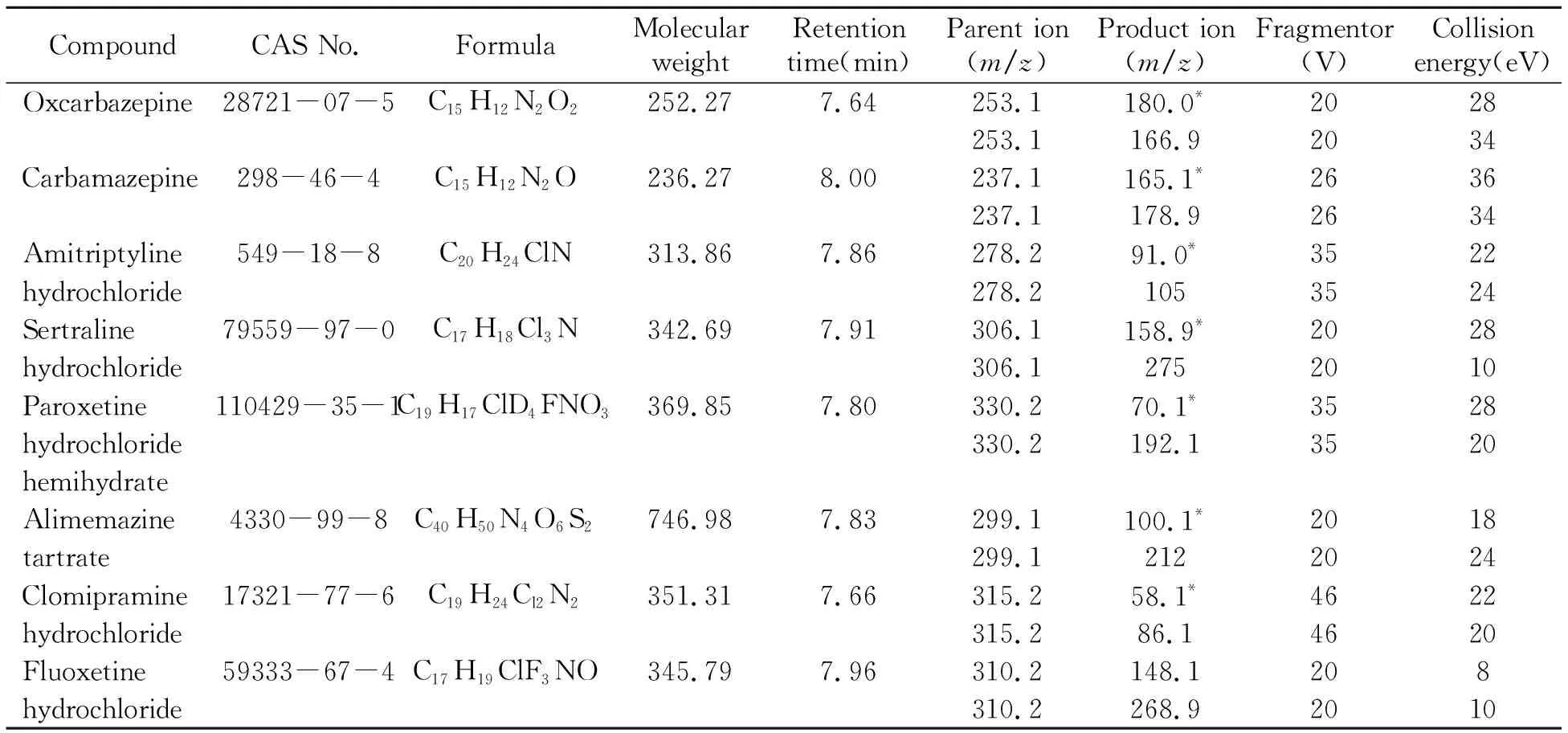
表1 精神类药物基本信息和质谱采集参数
1.2 样品前处理
水样经0.22 μm微孔滤膜过滤后用,甲酸调节pH值至3.0,将溶液转移至20 mL进样瓶中,在线固相萃取(Online SPE)装置吸取5 mL样品溶液进行净化富集,随后直接进行UPLC-MS/MS分析。
1.3 在线固相萃取条件
Oasis®HLB柱 (30×2.1 mm,20 μm);进样体积:5 mL;流动相A:1%甲酸水溶液;流动相B:甲醇/丙酮/正己烷(1/1/1,V/V/V)。Online SPE淋洗程序:0~0.5 min,2.00 mL/min,100%A;0.5~3.5 min,2.00~0.01 mL/min,100%A;3.5~4.5 min,0.01~2.00 mL/min,100%~0%A;4.5~7.5 min,0.01~2.00 mL/min,100%~0%A;7.5~13 min,2.00 mL/min,100%A。
1.4 超高效液相色谱条件
ACQUITY UPLCHSS T3色谱柱(100×2.1 mm,1.7 μm;Waters,USA);柱温:40 ℃;流动相A:0.5%甲酸水溶液,流动相B:乙腈;流速及梯度洗脱程序:0~4 min,0.1 mL/min,90%A;4~4.5 min,0.1~0.4 mL/min,90%A;4.5~9.5 min,0.4 mL/min,90%~10%A;9.5~10.5 min,0.4 mL/min,10%A;10.5~12 min,0.4 mL/min,10%~90%A;12~13 min,0.4 mL/min,90%A。
1.5 质谱条件
采用电喷雾正离子模式(ESI+)电离;多反应监测模式(MRM)监测;毛细管电压:1.00 kV;去溶剂气温度:500 ℃;去溶剂气流速:1 000 L/Hr;碰撞气流速:0.14 mL/min;离子源温度:150 ℃;雾化气压力:600 kPa。
1.6 样品数据处理
采用外标法定量,所有数据使用Waters MassLynx Software Ver.4.1(Waters Inc,USA)处理。
2 结果与讨论
2.1 样品基质的选择
饮用水所涵盖的领域非常广泛,并且有很多的分类。根据我国国家标准(GB 19298-2014)[16],选取饮用纯净水和其他饮用水两种基质。鉴于矿泉水管理方式的不同,取水地点、水源地保护、加工过程等有严格的特异性规定[17],将饮用天然矿泉水也加入到实验样品基质中。故最终确定实验的三种基质:饮用纯净水、饮用天然矿泉水和其他饮用水。
2.2 微孔滤膜的选择
为避免样品中的微小颗粒造成实验仪器管路的堵塞,实验中样品需先通过微孔滤膜进行过滤。分别考察了三种型号的0.22 μm微孔滤膜:PP、Nylon和GHP。向三种基质的阴性样品中添加8种目标化合物,浓度均为10 ng/L,分别通过上述三种型号的微孔滤膜。实验结果表明,Nylon膜过滤后有7种化合物的响应值是PP膜和GHP膜的1倍至2倍。故选用0.22 μm Nylon材质的微孔滤膜进行水样的过滤。
2.3 在线固相萃取条件的优化
2.3.1样液pH值和固相萃取柱的确定本实验采用Online SPE柱对样品中目标化合物进行富集,实验中考察了不同pH值(3、7和10)的样品溶液经过C18和HLB两种材料Online SPE后的富集效果。实验结果表明:上样液pH值为7和10时,两种SPE柱的提取效率在60%~120%之间的药物数量只有1~4种。在pH值为3时,8种目标化合物经过HLB柱后的回收率为68.9%~105.8%,但经C18柱富集后的目标化合物回收率均低于50%。故实验最终确定将样品调节至pH=3.0,并使用HLB柱作为本实验的Online SPE柱。
2.3.2淋洗液和洗脱液的优化Online SPE装置将样品注入SPE柱后,该装置流动相与SPE柱连接,淋洗液通过SPE柱进行淋洗。本实验中尝试在1%甲酸水溶液中加入5%的乙腈溶液,结果显示,8种目标化合物在质谱检测中没有出峰,说明淋洗液中的有机相比例过高,目标化合物在淋洗过程中无法保留在SPE柱上,故最终采用1%甲酸溶液水作为淋洗液。
SPE柱经过淋洗后,装置进行阀切换,将液相色谱流动相与SPE柱连接,该流动相将目标化合物反冲到色谱柱上进行分离,故SPE柱的洗脱液就是液相色谱的流动相溶液,其优化条件参见“2.4.2流动相的优化”。
2.4 色谱条件的优化
2.4.1色谱柱的选择样品经过富集后进入液相色谱柱进行进一步分离,为使8种目标化合物得到较好的峰形和分离效果,实验中分别比较了ACQUITY UPLC BEH C18色谱柱(100×2.1 mm,1.7 μm)和ACQUITY UPLC HSS T3色谱柱(100×2.1 mm,1.7 μm)两种色谱柱。结果表明:8种化合物经两种色谱柱分离后的色谱峰形对称,保留良好。从出峰时间比较,C18色谱柱上的保留时间较T3色谱柱提前0.1~0.4 min,8种化合物在T3色谱柱上保留较强。从响应值强度比较,T3色谱柱分离的化合物中有6种化合物的响应值是C18色谱柱的1至2倍。因此实验选择ACQUITY UPLC HSS T3色谱柱作为色谱分离柱。
2.4.2流动相的优化分别考察0.5%甲酸水溶液-乙腈、0.5%甲酸水溶液-甲醇、0.1%甲酸水溶液-乙腈和0.5%甲酸-0.1%乙酸铵水溶液-乙腈4种不同流动相条件下,8种化合物的出峰情况。实验结果表明:使用0.5%甲酸水溶液-甲醇作为流动相,只有2种目标化合物被检测出来,且定量限较高。其余三种流动相条件下均能将8种目标化合物洗脱出来,而在0.5%甲酸水溶液-乙腈流动相条件下,其响应值分别是其余两种流动相条件下的2至4倍左右。故选取0.5%甲酸水溶液-乙腈作为SPE净化富集和色谱分离的洗脱溶液。各物质的保留时间见表1。
2.5 质谱条件的确定
采用电喷雾(ESI)离子源,8种化合物均在正离子模式下电离,通过总离子流色谱图(图1)可以看出,8种化合物形成的准分子离子峰中[M+H]+峰丰度最高。参考欧盟2002/657/EC 指令[18],以准分子离子为母离子进行二级质谱扫描,选择丰度较高的两个碎片离子作为定量和定性离子,分别优化各个离子对的锥孔电压(Cone Voltage)及碰撞能量(Collision Energy)。优化去溶剂气温度及流量、碰撞气温度、雾化气压力、毛细管电压和离子源温度,使所有离子对获得最强响应信号。比较每对离子的强度和稳定性,最终选择响应较强的子离子作为定量子离子。8种目标化合物的质谱采集参数见表1。

图1 8种药物的总离子流(TIC)色谱图Fig.1 TIC chromatograms of 8 pharmaceuticals
2.6 线性范围和定量限
将8种目标物配制成0.1、0.2、0.5、5、10、20 ng/L 6个浓度梯度的混合标准溶液,分别得到对应物质的标准曲线,通过向没有目标物的样品基质中添加标准物质,按10倍信噪比进行定量限(LOQ)的计算,每种化合物的标准曲线方程、线性范围、相关系数和LOQ见表2。由表2可看出,这8种化合物在线性范围内有良好的线性关系,相关系数(R)均大于0.995,灵敏度较高。
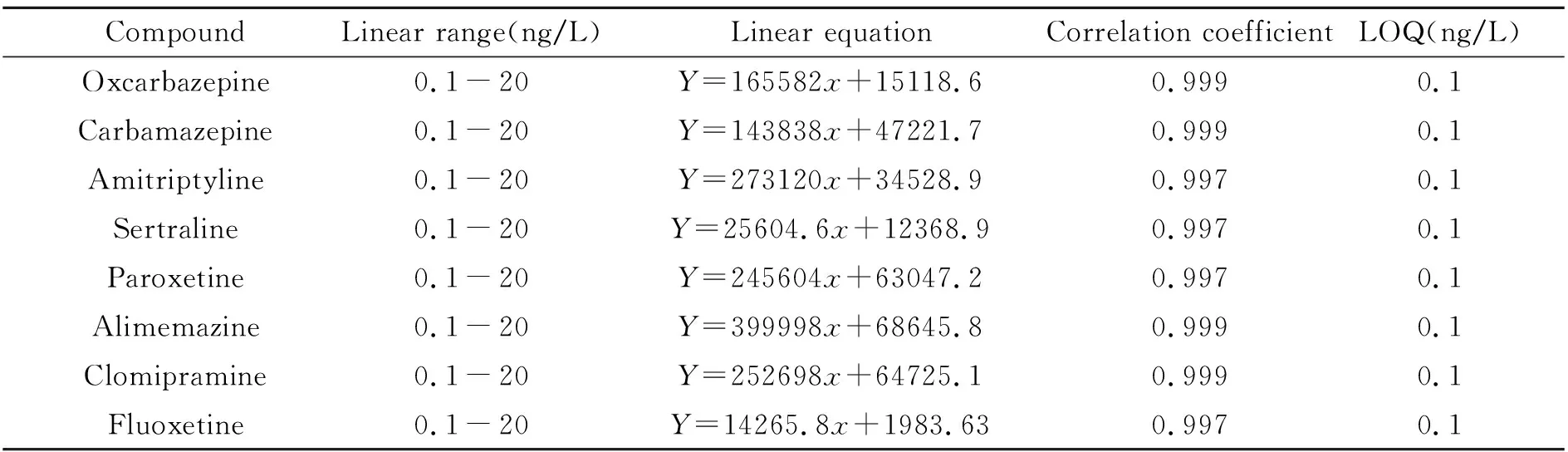
表2 8种药物的线性范围、线性方程、相关系数和定量限(LOQ)
2.7 方法的准确度与精密度
对饮用纯净水、其他饮用水和饮用天然矿泉水三种基质阴性样品分别进行了8种化合物的添加回收实验,分别选取0.1 ng/L和1 ng/L作为加标水平,每个添加水平进行6次平行实验,外标法定量,计算得到平均回收率和相对标准偏差(RSD),实验结果见表3。从表3可以看出,在选定的2个水平下,添加回收率在68.9%~105.8%之间,RSD在3.6%~13.6%之间,满足方法学的要求。
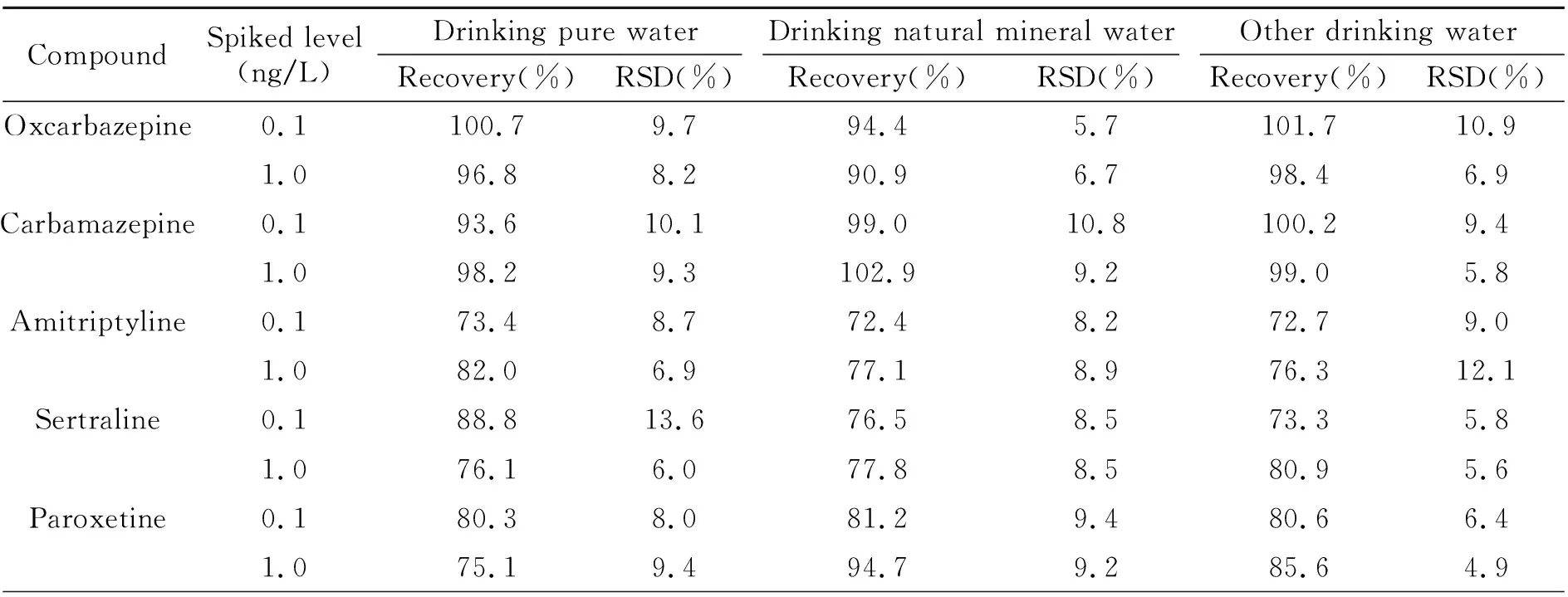
表3 饮用纯净水、其他饮用水和饮用天然矿泉水中8种药物加入回收率及相对标准偏差(RSD)(n=6)
(续表3)
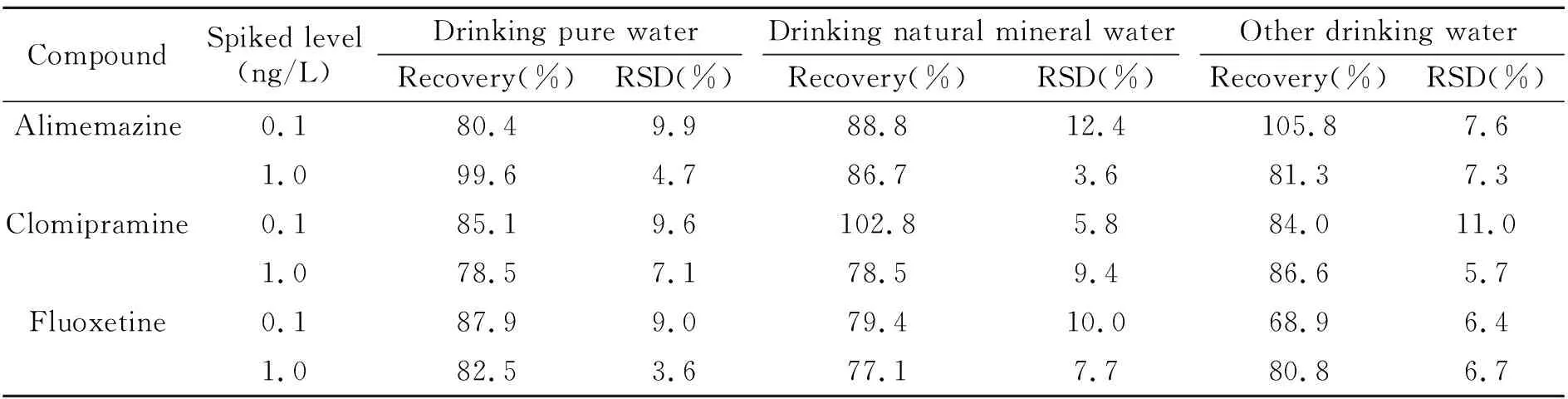
CompoundSpiked level(ng/L)Drinking pure waterDrinking natural mineral waterOther drinking waterRecovery(%)RSD(%)Recovery(%)RSD(%)Recovery(%)RSD(%)Alimemazine0.180.49.988.812.4105.87.61.099.64.786.73.681.37.3Clomipramine0.185.19.6102.85.884.011.01.078.57.178.59.486.65.7Fluoxetine0.187.99.079.410.068.96.41.082.53.677.17.780.86.7
2.8 实际水样的测定
应用本实验方法对北京市部分饮用水源地采集的39份水样进行8种目标物残留检测。结果显示,在39份水样中有8份样品检测出奥卡西平,浓度范围为1.4~8.2 ng/L;有14份样品检测出卡马西平,浓度范围为0.21~4.6 ng/L,其余6种目标物测定结果低于检出限。标准品和样品中卡马西平的MRM参考色谱图见图2。

图2 卡马西平标准品(a)和样品中卡马西平(b)的MRM色谱图Fig.2 MRM chromatograms of carbamazepine standard(a) and carbamazepine in sample(b)
3 结论
本研究建立了饮用水中8种常见精神类药物的在线固相萃取-超高效液相色谱/串联质谱测定方法。该方法简便快捷,节省人力,满足灵敏度和精密度的要求。应用本方法对北京市饮用水源水样品进行药物残留分析,8份样品检测出奥卡西平,14份样品检测出卡马西平。这些测定数据的积累可以为今后饮用水水质安全风险评估和法规制定提供必要的技术支撑。
