Specific genetic variation in two non-motile substrains of the model cyanobacterium Synechocystis sp. PCC 6803*
2018-12-22CHENJun陈军SHIWenjun史文军LIWenjun李文军CHENGao陈高QINSong秦松
CHEN Jun (陈军) , SHI Wenjun (史文军) , LI Wenjun (李文军) CHEN Gao (陈高) , QIN Song (秦松)
1 Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, Yantai 264003, China
2 University of Chinese Academy of Sciences, Beijing 100049, China
3 Biotechnology Research Centre, Shandong Academy of Agricultural Sciences, Jinan 250100, China
Abstract Synechocystis sp. PCC 6803 is a model organism widely used in cyanobacterium biology and biotechnology. To know the genetic background of substrains of Synechocystis sp. PCC 6803 is important for further research and application. In this study, we reported the genome sequences of two non-motile wild-type substrains of Synechocystis sp. PCC 6803 using whole genome resequencing. 55/56 putative single nucleotide polymorphisms (SNPs) and 8/9 Indels (insertion and deletion) were identified. Among these, 47 SNPs were found in both the GT-AR and GT-CH strains, and 8 were unique to GT-AR and 9 were unique to GT-CH. All of these variations were annotated in metabolism pathway referred to KEGG database. Meanwhile, the deletion in slr0332 gene was detected in these two strains, which attributed to the non-motile phenotype of them and suggested that the insertion in spkA gene was not essential for non-motile phenotype. These resequencing data provide the genetic background information of these two strains and highlighted the microevolution over decades of laboratory cultivation.
Keyword: Synechocystis sp. PCC 6803; genome resequencing; non-motile; genetic background
1 INTRODUCTION
Due to the unsustainable consumption of fossil fuels and rapidly increased energy needs, to develop sustainable bioenergy has been an urgent demand.Cyanobacteria, which could use sunlight and CO2as carbon sources to produce organic materials, have received considerable interests as a sustainable energy resource (Dismukes et al., 2008; Melis, 2009). The freshwater speciesSynechocystissp. PCC 6803 is a model cyanobacterium which has many excellent characteristics for biofuels production: I) it is naturally transformable by incorporating exogenous DNA into cells that is integrated into the genome with high frequency, which make it easy to be constructed for biofuel production by genetic modified or metabolic engineering methods (Williams, 1988); (II) it is capable to grow under photoautotrophic, mixotrophic,and heterotrophic conditions (Rippka et al., 1979;Williams, 1988; Anderson and McIntosh, 1991),while the presence of glucose can significantly increase biomass and high-value bioproducts production (Yoshikawa et al., 2013; Nakajima et al.,2014); III)Synechocystissp. PCC 6803 was the first cyanobacteria to have its whole genome sequenced,which promoted the postgenomic research such as transcriptome, proteome and functional genomic studies (Ikeuchi and Tabata, 2001). Nowadays,Synechocystissp. PCC 6803 is widely used as a cell factory for bioethanol (Dexter and Fu, 2009),hydrogen (McCormick et al., 2013), isobutanol(Varman et al., 2013), fatty alcohols and hydrocarbons production (Tan et al., 2011) by metabolic engineering methods.
Information about the precise genetic background of the usedSynechocystissp. PCC 6803 strains is essential for the intended construction of robust biofuel producer strains. The original strain ofSynechocystissp. PCC 6803 was named Berkeley strain, isolated from California freshwater by Kunisawas and colleagues (Stanier et al., 1971). Then it was deposited in the Pasteur Culture Collection(PCC strain) and the American Type Culture Collection (ATCC strain). The glucose-tolerant strain(GT) was subsequently isolated from the ATCC strain(Rippka et al., 1979). However, these substrains have different phenotypes and genetic backgrounds. To analysis these different variations, a series ofSynechocystissp. PCC 6803 strains’ genomes were sequenced. The genome ofSynechocystissp. PCC 6803 GT-Kazusa strain, derived from GT-strain,consisted of a single chromosome and several plasmids in different sizes. The single chromosome and three plasmids were sequenced and 3 168 potential genes were predicted, which established a hallmark in the study of cyanobacteria (Yang and McFadden, 1993, 1994; Kaneko et al., 1996; Xu and McFadden, 1997). Kaneko et al. (2003) sequenced another four large plasmids (pSYSM: 120 kb,pSYSX: 106 kb, pSYSA: 103 kb and pSYSG: 44 kb)(Kaneko et al., 2003). The genome structure of GT-S strain, derived from GT-Kazusa strain, was analyzed and 22 putative single nucleotide polymorphisms(SNPs) errors of GT-Kazusa genome sequence were detected by comparing the genome sequence of the strain GT-S with GT-Kazusa database (Tajima et al.,2011).
To establish reliable genomic sequence data and investigate microevolution processes inSynechocystissp. PCC 6803, the genomes of three substrains (GT-I,PCC-P and PCC-N) were resequenced by massively parallel whole-genome resequencing (Kanesaki et al.,2012). Of these substrains, GT-I was isolated as a single colony from the GT-strain by Ikeuchi and Tabata (Ikeuchi and Tabata, 2001; Kanesaki et al.,2012). The substrains PCC-P and PCC-N were isolated from the PCC strain and designated according to the direction of phototactic movement (Yoshihara et al., 2001). Trautmann et al. (2012) re-sequenced a motile and glucose tolerant substrain PCC-M, which revealed that the considerable evidence for the trace of microevolution during cultivation in the laboratory(Trautmann et al., 2012).
In this study, to clarify the genomic background of two non-motile wild-type strains ofSynechocystissp.PCC 6803 maintained in our lab, the genetic variation of them were identified by whole genome resequencing. And the genes affected by SNP were also annotated against KEGG database. These results will provide us important information about these two strains on genetic variation for further research and application, such as building the mutants for biofuel production based on these strains. They also suggested us that the genetic variation was widely distributed in the genome among differentSynechocystis6803 strains and presented its evolution and divergence globally.
2 MATERIAL AND METHOD
2.1 Algal strain and DNA extraction
The strains GT-AR and GT-CH were maintained in Biotechnology Research Center, Shandong Academy of Agricultural Sciences. The strains were cultured in BG11 medium inoculated at 5% (v/v) into 100 mL flasks with illumination of 40 μmol photons/(m2·s) at 28°C. After they were grown to late exponential phase, they were used as seed cultures for the following experiments. The seed cultures were harvested, centrifuged and aliquoted into 500 mL flasks with 200 mL BG11 medium, then cultured at 28°C on an orbital shaker (120 r/min)with 40 μmol photons/(m2·s). The strains in exponential growth were harvested by centrifugation for 12 000×g, 10 min at 4°C, and rinsed with PBS.DNA was extracted following the protocol described by Sambrook and Russell (2000) (Sambrook and Russell, 2000). Total DNA obtained was subjected to quality control by agarose gel electrophoresis and determined by NanoDrop 2000c (Thermo Scientific,USA).
2.2 Sequencing methods and data analysis
The genome was sequenced with MPS (massively parallel sequencing) method by the platform illumina HiSeq 4000. A paired-end library with an insert size of 350 bp was constructed using an Illumina HiSeq 4000 by PE150 strategy. The sequence of adapter and low-quality data were removed, resulting in the clean data used for subsequent analysis. The clean data were mapped to the GT-Kazusa reference genome(Accession number: NC_000911.1) using BWA software (Li and Durbin, 2009). Then the coverage of the reference sequence to the reads and the explanations of the alignment results were investigated using SAMTOOLS software (Li et al., 2009). The detection of the individual SNP and insertion and deletion of small fragments (<50 bp), as well as the variation of SNP/InDel in the functional regions of the genome were also investigated using SAMTOOLS software (Li et al., 2009). The insertion (INS) and deletion (DEL) between these two samples and reference genomes were detected by BreakDancer software (Chen et al., 2009). The variation map of the whole genome was created by Circos to show reads coverage and the distribution of SNP and InDel information (Krzywinski et al., 2009). Gene mutations annotation and classification were performed against KEGG database using diamond software (Buchfink et al., 2015).
3 RESULT AND DISCUSSION
3.1 Genome resequencing and data overview
In this study, 877.31/879.14 Mbp raw data of GTAR and GT-CH were obtained, and 806/805 Mbp clean data of them were summarized, respectively.The sequencing average-depth was 138/132-fold and its coverage >99.99. The total base of sample GT-AR and GT-CH were 543 432 600 and 522 600 900 bp.The map rates of clean data were about 90% compared with the reference genome GT-Kazusa.
3.2 SNPs and indels detection and functional annotation
We reported a total of 55/56 SNPs in the chromosome of GT-AR and GT-CH compared with the database records for GT-Kazusa. Of these SNPs,21/21 are synonymous mutations, 25/27 substitutions lead to amino acid substitutions, and 9/8 are intergenic mutations. Then 47 SNPs were found in both the GTAR and GT-CH (Table 1). Among these, 15 mutations have been reported previously, including 9 database errors (#4, #5, #7, #8, #13, #20, #21, #22, #29) of GTKazusa (Tajima et al., 2011), as shown in Table 1 with asterisk-marked. Mutation #1 occurred inslr1609gene, which encoded long-chain fatty acid-CoA ligase. The gene played crucial role for production of free fatty acids and fatty acid derivatives metabolically(Gao et al., 2012). Thus it may affect the fatty acids composition compared with other strains. Mutation#2 occurred insll1968gene encoding photomixotrophic growth-like protein (pmgA), which was essential for photomixotrophic growth, whereas it repressed photoautotrophic growth (Hihara and Ikeuchi, 1997; Takahashi et al., 2008). Seven mutations (#3, #6, #9, #10, #11, #25, and #26) may affect the function ofslr1682,sll0092,slr0511,ssl0172andslr1684genes encoding transposase.Mutation #12 occurred insll1625gene encoding(2Fe-2S)-binding protein. Mutation #27 occurred insll0550gene, and the gene product was diflavin flavoprotein A which shared with the GT-O2 strain(Morris et al., 2014). The gene played important role in Mehler reaction (photoreduction of O2to H2O) as a NAD(P)H oxygen oxidoreductase (Allahverdiyeva et al., 2011).
Mutation #28 occurred insll2012gene encoding RNA polymerase sigma factor RpoD/SigA family. InSynechocystissp. PCC 6803, nine species of sigma factor homologues have been identified based on amino acid sequence: group 1 (SigAgene product,SigA), group 2 (SigB,SigC,SigD, andSigE) and group 3 (SigF,SigG,SigHandSigI), respectively.SigAmay be a principal sigma factor.SigBandSigDwere heat-shock and high-light responsive sigma factors, respectively (Imamura et al., 2003).SigFgene played a critical role in motility via the control of pili formation and was likely to regulate other features of the cell surface (Bhaya et al., 1999). Three mutations (#34, #35 and #36) appeared inslr0665gene described as bifunctional aconitate hydratase 2/2-methylisocitrate dehydratase, which may also affect the tricarboxylic acid cycle. Mutation #37 occurred inslr0335gene encoding phycobiliprotein ApcE, which was large anchor protein LCM appeared to be a central component in attachment of the phycobilisome to the thylakoid membrane and in the transfer of excitation energy from phycobilins to chlorophyll in the thylakoid.
Besides, twelve mutations (#14, #15, #16, #17, #18,#19, #23, #24, #30, #31, #32, #33) appeared insll1774,slr1712,slr1683andslr1056genes, and these gene products were hypothetical protein, the function of which were still remained to be characterized. Eight mutations (#40, #41, #42, #43, #44, #45, #46, #47)were detected in non-coding region and the function of them were also not clearly. Compared with the GTKazuse genome sequences, 8 SNPs unique to GT-AR and 9 SNPs unique to GT-CH were detected. The SNPs appeared mainly in genes encoding transposase(#57, #58, #59, and #60) and hypothetical protein(#56, #61, #62, #63, and #64) in GT-CH strain. As unique to GT-AR strain, the SNPs also mainly occurred in transposase (#48, #49) and hypothetical protein(#50, #51, #52, #53). Besides, the gene encoding aconitate hydratase B had another mutation (#54), thus the tricarboxylic acid cycle may be different between GT-AR and GT-CH strains (Table 1).

Table 1Location and effects of SNPs identified in the chromosome of GT-ARand GT-CH comparedwith the database records for GT-Kazusa

Table 1 Continued
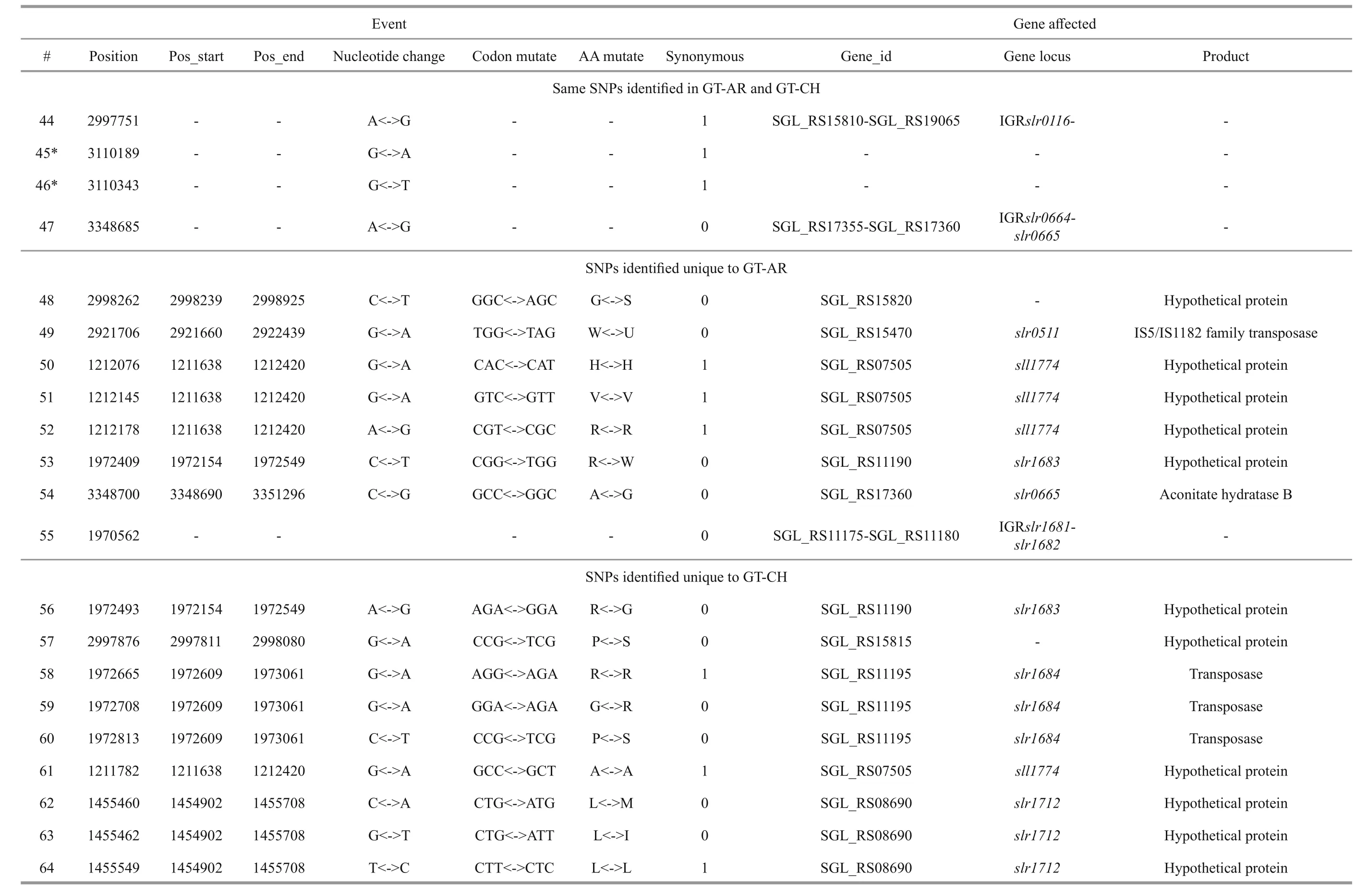
Table 1 Continued

Table 2 Location and effects of indels identified in GT-AR and GT-CH compared with the database records for GT-Kazusa
Indels were described as the short reads less than 50 bp which were inserted or deleted in the genome,called “insertions and deletions”. In this study, the Indels in the genome of GT-AR and GT-CH were identified by comparing the aligned reads to reference assembly genomes GT-Kazusa. The results indicated that 8/9 Indels were detected for the strain,respectively. Of these, there are 4/5 insertions. On the other hand, 4/4 deletions were identified in GT-AR and GT-CH against the referred genome GT-Kazusa.
Among these Indels, 8 were found in both the GTAR and GT-CH strains, including four insertions(position 967340, 2350286, 2360247, 2544045) and four deletions (position 2204584, 2409244, 2419399,3260096). Besides, the insertion (position 1427498)unique to GT-AR was detected. As shown in Table 2,gene affected by the deletion at position 2204584 against referred genome encoded pilus assembly protein pilC, which was a cytoplasmic membrane polypeptide required for pilus assemble. Bhaya et al.(2000) sequenced the gene region and found a single base difference with respect to the reported sequence between positions 2204576 and 2204584, and proposed that the lack of motility ofSynechocystissp.PCC6803 strain may be due to the putative frameshift mutation inpilC(Bhaya et al., 2000). This suggested us that these two strains could not motile in this study due to the deletion at position 2204584 in the genome.
In general, the Berkeley and PCC strains are motile,while the others are apparently non-motile, which could be distinguished by sequencingspkA,pilCandslr2031(Ikeuchi and Tabata, 2001). Ding et al. (2015)identified that the 1 bp insertion inspkAand the deletion inpilCwere attributed to the non-motile phenotype of GT-G strain (Ding et al., 2015). But the impact of individual mutations of these two genes is still not clear. In this study, we found the deletion inpilC, but not detected the mutation inspkA. Thus we deduced that the individual mutation ofpilCcould lead to change the phenotype.
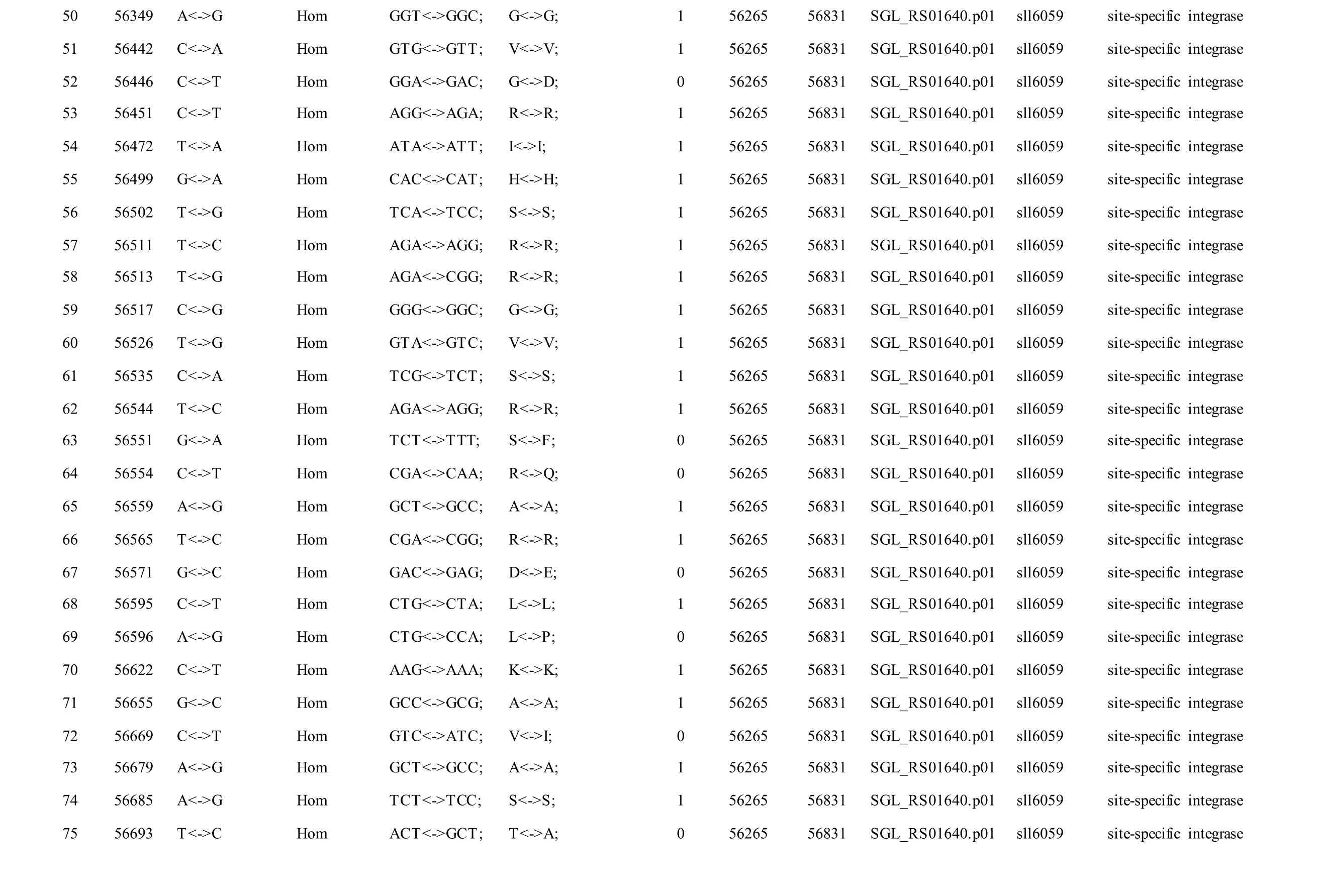
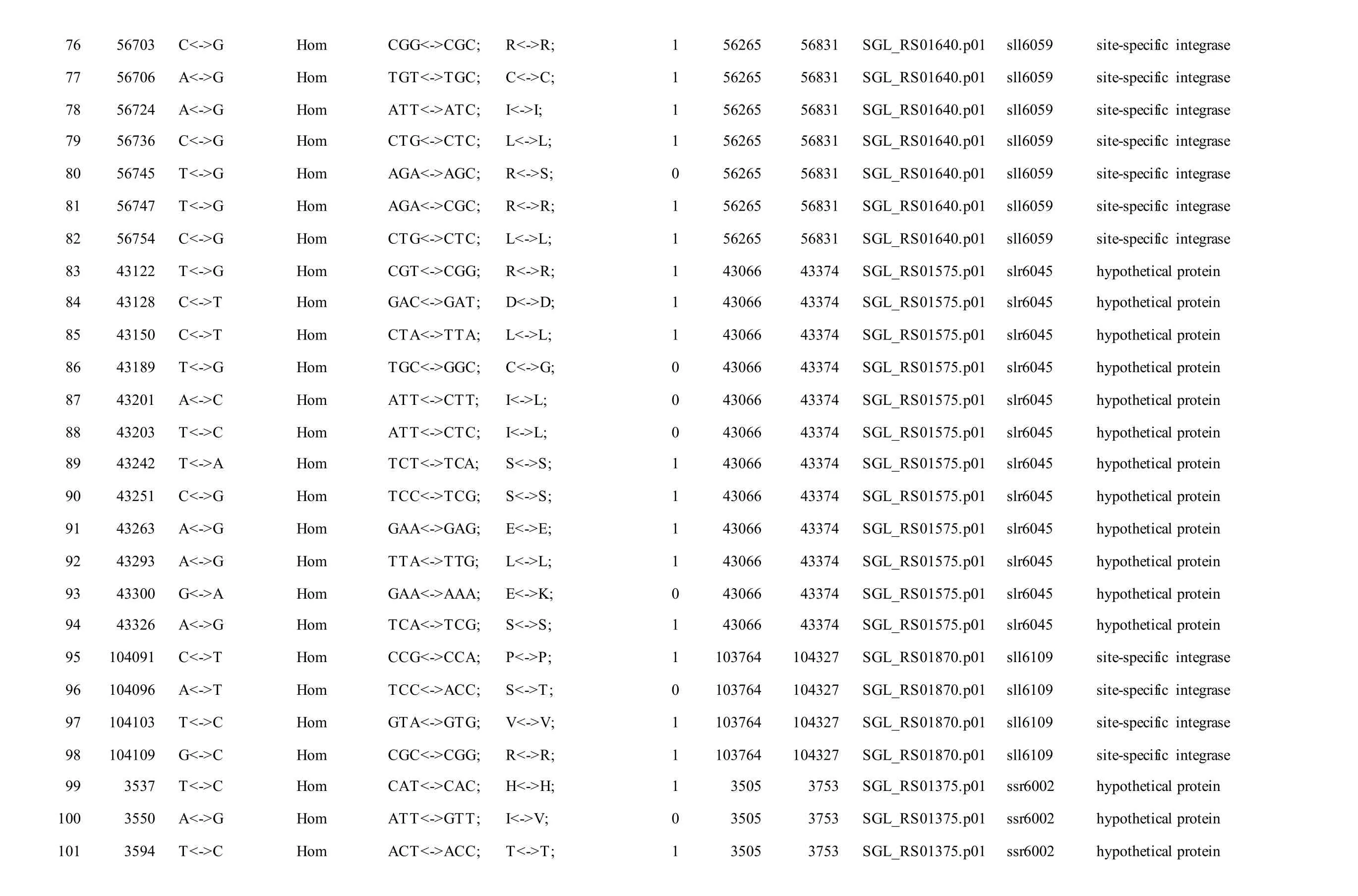
Meanwhile, several SNPs and Indels were also detected in the plasmids of GT-AR and GT-CH compared with the database records for GT-Kazusa(Supplementary file). Among these seven plasmids,the SNPs and Indels were mainly involved in plasmid pSYSX, pSYSM and PSYSA. The mutations identified in pSYSX of both GT-AR and GT-CH occurred in genesslr6040,slr6070,slr6037,ssr6062,sll6059,slr6045,sll6109,ssr6002andslr6039, which encoded DNA-binding response regulator, twocomponent sensor histidine kinase, arsenate reductase ArsI2, hypothetical protein, site-specific integrase,hypothetical protein, site-specific integrase,hypothetical protein and DUF305 domain-containing protein, respectively. The genes affected by SNPs identified unique to GT-CH weresll6059,ssr6089,sll6059andslr6045encoding two-component sensor histidine kinase, HNH endonuclease and arsenate reductase ArsI2, respectively, while the genesslr6038andssr6032encoded hypothetical protein. For another, the genes affected by SNPs identified unique to GT-AR weresll6109,slr6094,ssr6002andslr6038encoding hypothetical protein (Table S1).
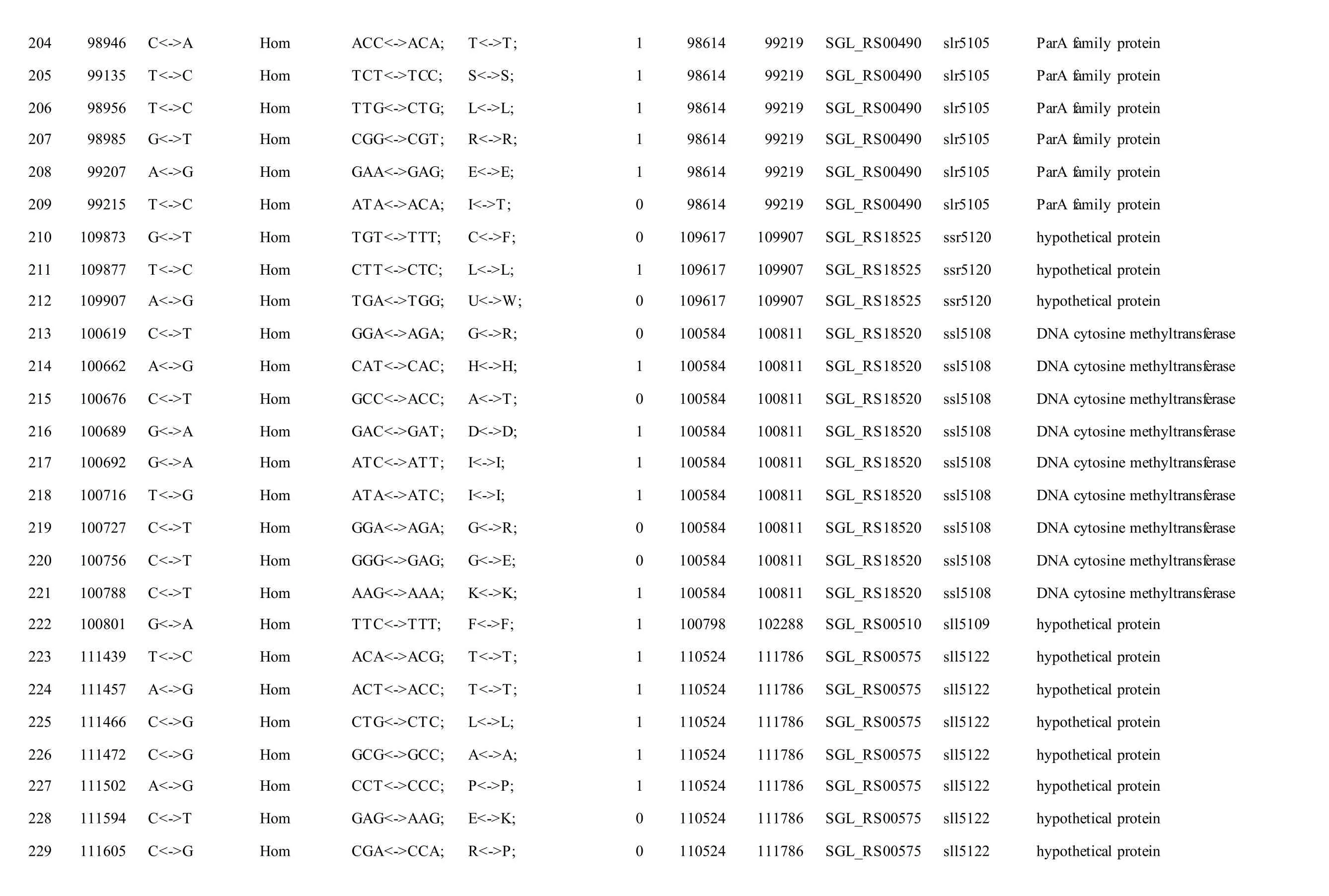
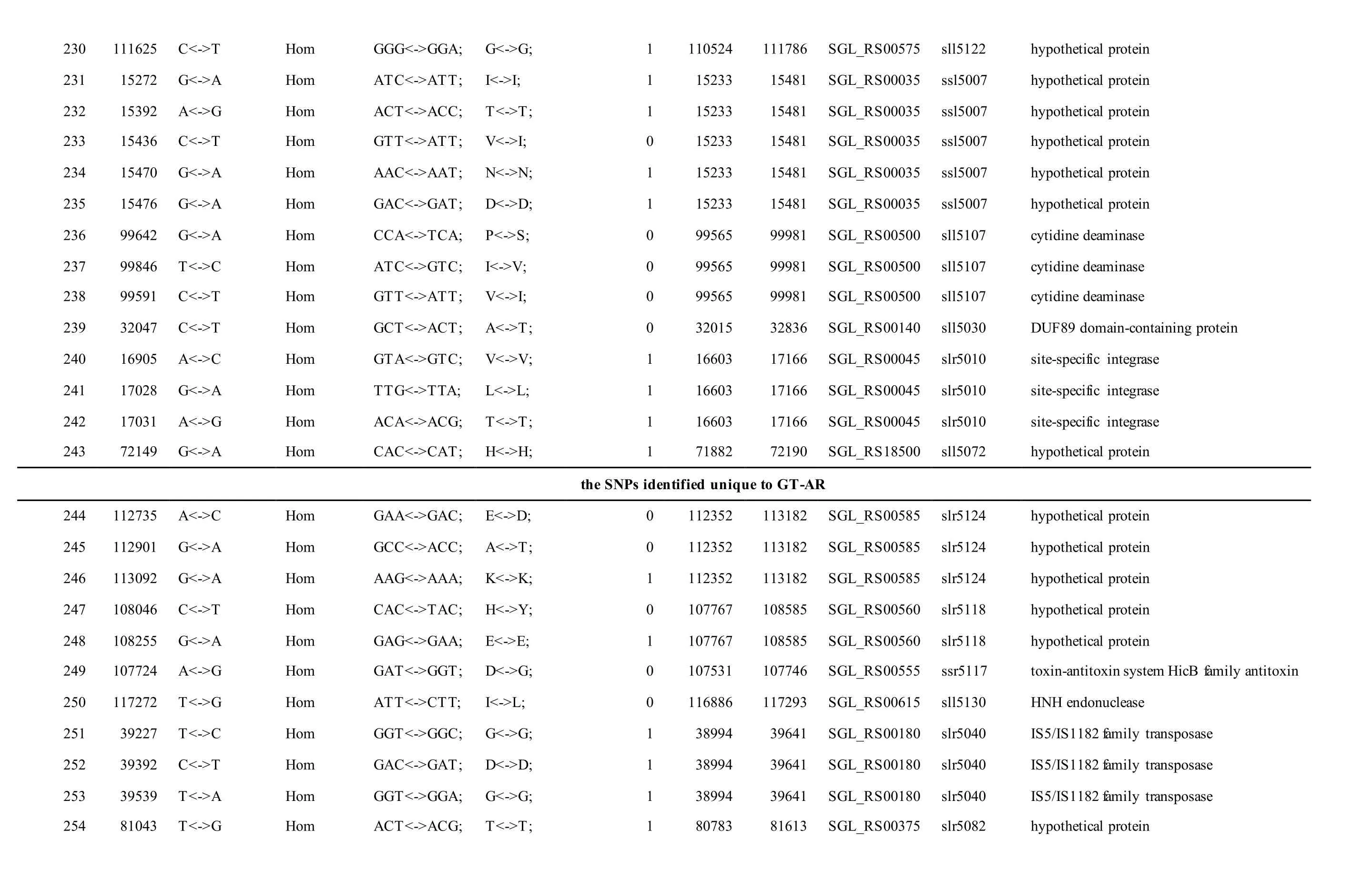
The mutations identified in pSYSM of both GTAR and GT-CH occurred in genesslr5029,ssr5117,ssl5108,slr5010,sll5089,slr5105,ssl5108,sll5107,sll5030andslr5010, which encoded IS701 family transposase, toxin-antitoxin system HicB family antitoxin, DNA cytosine methyltransferase, sitespecific integrase, metal-dependent hydrolase, ParA family protein, DNA cytosine methyltransferase,cytidine deaminase, DUF89 domain-containing protein and site-specific integrase, respectively.Several genes includingslr5124,sll5084,slr5118,sll5090,sll5006,ssl5091,slr5082,ssr5121,slr5119,sll5072,slr5077,ssl5108,ssr5106,ssl5068,ssr5120,sll5109,sll5122,ssl5007andsll5072encoded hypothetical protein, the function of which were still remained to be verified. The genes affected by SNPs identified unique to GT-AR weressr5117,sll5130,slr5040,sll5089,slr5124,slr5118,slr5082,slr5077,slr5119,sll5072. Among these, the genesssr5117,sll5130,slr5040andsll5089encoded toxin-antitoxin system HicB family antitoxin, HNH endonuclease,IS5/IS1182 family transposase and metal-dependent hydrolase, repectively, while the function of genesslr5124,slr5118,slr5082,slr5077,slr5119,sll5072encoded hypothetical protein (Table S2). Besides,eight Indels including six deletions and two insertions were detected in both GT-AR and GT-CH (Table S3).
The mutations identified in pSYSA of both GT-AR and GT-CH occurred in genessll7002,slr7005andsll7001encoding IS701 family transposase, sitespecific integrase and hypothetical protein,respectively. Several genes includingslr5124,sll5084,slr5118,sll5090,sll5006,ssl5091,slr5082,ssr5121,slr5119,sll5072,slr5077,ssl5108,ssr5106,ssl5068,ssr5120,sll5109,sll5122,ssl5007andsll5072encoded hypothetical protein, the function of which were still remained to be verified. Besides,some specific mutations were identified in the genesslr7005andsll7001in these two different strains(Table S4). Three Indels identified in both GT-AR and GT-CH were at positions 376, 721 and 850, and may affect the genessll7001andsll7002encoding hypothetical protein and IS701 family transposase,respectively (Table S5).
In total, compared with the referred genome, the result indicated that the genomic sequence for both these two non-motile strains were relatively unique,sharing 55/56 SNPs and 8/9 Indels in GT-AR and GTCH. And only 15 SNPs were previously reported,including 9 errors of the original GT-Kazusa sequence.For another, seven Indels (position 1427498, 2204584,2350286, 2409244, 2419399, 2544045, 3260096) are previously reported, which were asterisk-marked at Table 2. Of these, five Indels (position 2350286,2409244, 2419399, 2544045, 3260096) shared with the finished genomes of GT-Kazusa, GT-S, GT-I, GTO1, GT-O2, GT-G, PCC-P, PCC-N, PCC-M. The Indel (position 2204584) also shared with the finished genomes of GT-S, GT-I, GT-O1, GT-O2, GT-G,PCC-P, PCC-N, PCC-M, but not shared with the genome of GT-Kazusa (Morris et al., 2014). These results suggested that microevolution process existed in these two strains during laboratories maintenance.
3.3 Gene mutations classification
Furthermore, to identify and analysis the active biological pathway which may be affected by SNPs,these annotated SNPs were mapped against the reference biochemical pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG)database. KEGG is a database resource for understanding high-level functions and utilities of the biological system from molecular-level information,including KEGG pathway database, KEGG orthology database and other 14 databases (http://www.genome.jp/kegg/). As group A, group B, group C and group D shown in Fig.1, annotated against the KEGG pathway database (not include human disease pathway), 1,011 genes in the reference genome of GT-Kazusa strain were mapped in 17 pathways. 14 mutated genes found in both GT-AR and GT-CH have been classified into group E, mainly involved in nitrogen metabolism,carbon fixation, butanoate metabolism, glyoxylate and dicarboxylate metabolism, toluene degradation, starch and sucrose metabolism, cyanoamino acid metabolism,alanine aspartate and glutamate metabolism,pyrimidine metabolism, oxidative phosphorylation,fatty acid metabolism and citrate cycle (TCA cycle)(Fig.1). Besides, these SNPs unique to GT-AR were classified in the group F as showed in Fig.1, three pathways were involved in these gene mutations,including carbon fixation, glyoxylate and dicarboxylate metabolism and citrate cycle (TCA cycle). The mutations unique to GT-CH were also mapped in KEGG, but no genes were annotated, which may be due to the mutated genes were mainly transposase and hypothetical protein. Thus, the difference between these two strains were mainly involved in carbon metabolism, which should be taken into consideration when performing mutants constructing or other phenotypic analysis in further research.
4 CONCLUSION
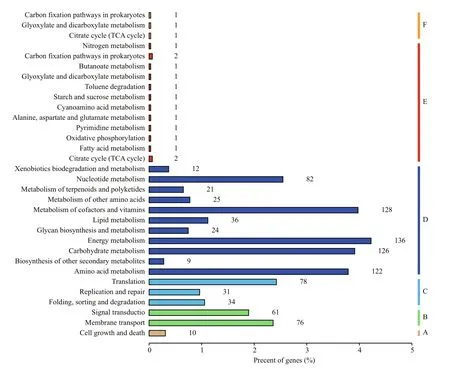
Fig.1 Functional categories of genes affected by SNPs in GT-AR and GT-CH
55/56 SNPs and 8/9 indels were detected in GT-AR and GT-CH strains ofSynechocystissp. PCC 6803 using whole genome resequencing method. Only 15/15 SNPs and 6/6 indels were previously reported,including 9 sequencing errors in the genome of GTKazusa. The result suggested us that these two strains GT-AR and GT-CH were relatively unique to the previous reported strains and microevolution occurred in them during laboratory maintenance. Then the mutation inslr0332gene was found in these two strains, which attributed to their non-motile phenotype. This fact gave us evidence that the deletion inslr0332could lead to the phenotype change,although the insertion inspkAwas not existed.Meanwhile, by annotating and classifying genes may affect by SNPs against KEGG database, we found the differences between these two strains were mainly involved in carbon metabolism. These provided important information for us to know the background of these strains maintained in our laboratory, and highlighted need to select a suitable strain to construct mutants for biofuel and chemicals production in further research. Meanwhile, the data will be useful for further investigating the evolution and divergence ofSynechocystisin laboratories worldwide.
Electronic supplementary material
Supplementary material (Supplementary Table S1–S5) is available in the online version of this article at https://doi.org/10.1007/s00343-019-7291-1.
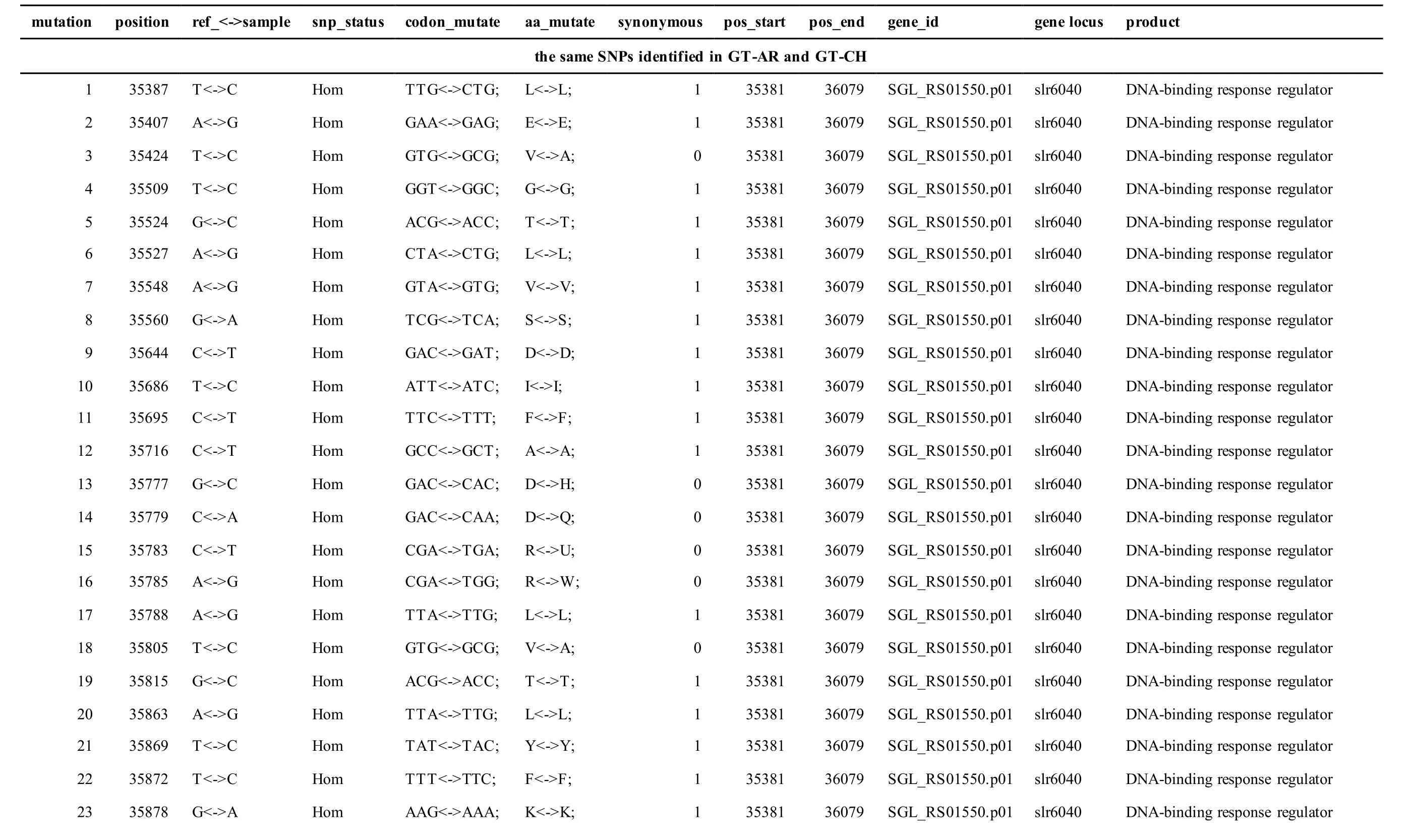
Table S1 Location and effects of SNPs identified in the plasmid pSYSX of GT-AR and GT-CH compared with the database records for GT-Kazusa.

?
?
?

?

?

Table S2 Location and effects of SNPs identified in the plasmid pSYSM of GT-AR and GT-CH compared with the database records for GT-Kazusa

?

?

?

?

?

?

?
?
?

?

?

Table S3 Location and effects of Indels identified in the plasmid pSYSM of GT-AR and GT-CH compared with the database records for GT-Kazusa
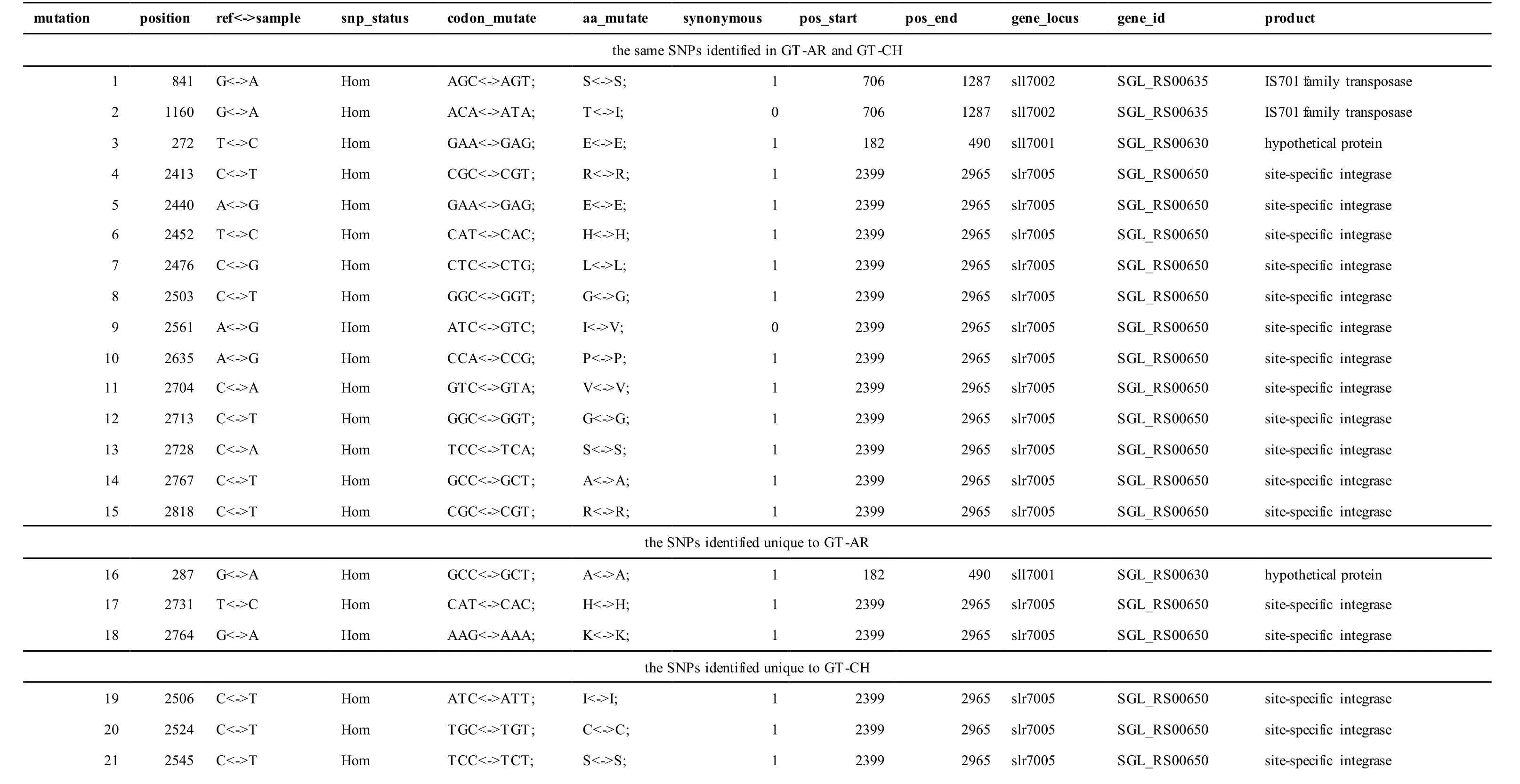
Table S4 Location and effects of SNPs identified in the plasmid pSYSA of GT-AR and GT-CH compared with the database records for GT-Kazusa

?
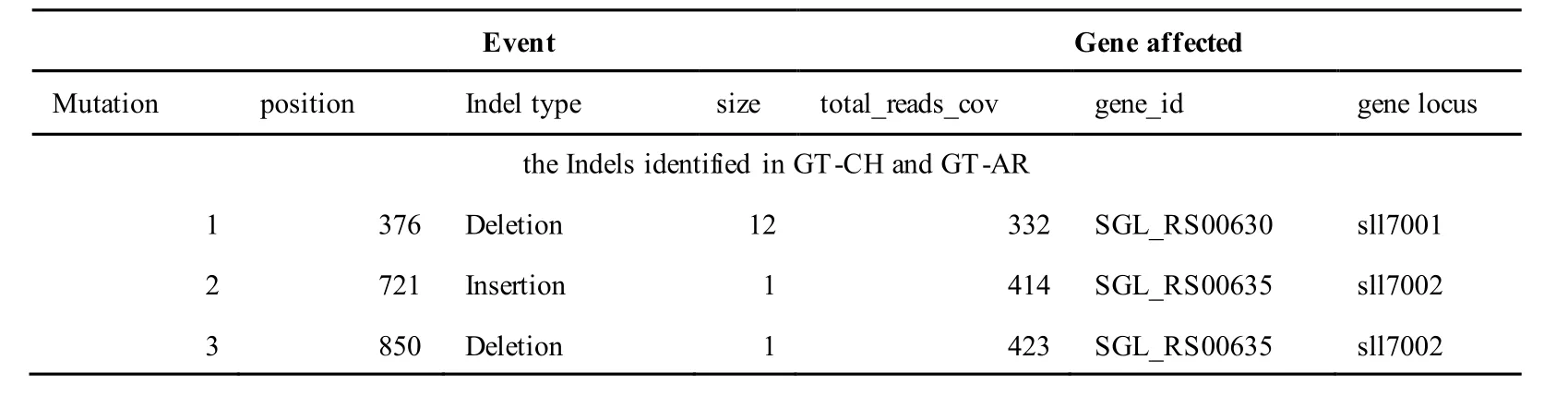
Table S5 Location and effects of Indels identified in the plasmid pSYSM of GT-AR and GT-CH compared with the database records for GT-Kazusa
猜你喜欢
杂志排行

Journal of Oceanology and Limnology的其它文章
- Neuroanatomy and morphological diversity of brain cells from adult crayfish Cherax quadricarinatus*
- Effects of feeding time on complement component C7 expression in Pelteobagrus vachellii subject to bacterial challenge*
- Cryopreservation of strip spawned sperm using programmable freezing technique in the blue mussel Mytilus galloprovincialis*
- Pf- D mrt4, a potential factor in sexual development in the pearl oyster Pinctada f ucata*
- Functional characterization of a Δ6 fatty acid desaturase gene and its 5′-upstream region cloned from the arachidonic acidrich microalga Myrmecia incisa Reisigl (Chlorophyta)*
- The expression characteristics of vitellogenin (VTG)in response to B(a)p exposure in polychaete Perinereis aibuhitensis*