MnO2催化O3处理准好氧矿化垃圾床渗滤液尾水中难降解有机物
2018-11-28邓禺南陈炜鸣罗梓尹崔瑜旗李启彬
邓禺南,陈炜鸣,罗梓尹,崔瑜旗,李启彬
MnO2催化O3处理准好氧矿化垃圾床渗滤液尾水中难降解有机物
邓禺南,陈炜鸣,罗梓尹,崔瑜旗,李启彬*
(西南交通大学地球科学与环境工程学院,四川 成都 611756)
研究利用MnO2催化O3氧化技术对准好氧矿化垃圾床渗滤液尾水中难降解有机物进行处理.系统研究了臭氧投量、二氧化锰投量和初始pH值对有机物的去除效果和反应动力学.通过UV-Vis和3D-EEM技术对渗滤液尾水中溶解性有机物分子结构在O3/MnO2体系的转化进行了探究,在此基础上,运用XRD、SEM、EDS和XPS对二氧化锰的催化机理进行研究.结果表明:在反应时间为20min,O3投量为18.92mg/min,初始pH值为3时,添加0.2g/L的MnO2能显著提升有机污染物的去除能力,较单独O3作用,其COD、UV254、色度 CN 的去除率分别提升了24.66%、4.95%和12.57%,并且生色团的有机物最易被臭氧氧化降解.紫外-可见光谱和三维荧光显示,O3/MnO2体系能使废水中腐殖质降解,其有机物的芳香性程度、分子量和缩合度均降低,体系对苯环类化合物的降解效率提高,最终,废水的可生化性得到显著提高.与此同时,反应前后MnO2未出现新增价态峰值的变化,Mn(Ⅳ)在催化过程中占主导地位,MnO2在O3/MnO2体系中协同机理为MnO2催化O3产生羟基自由基以及MnO2在体系中转化为水合二氧化锰,改变催化剂表面理化性质与有机物形成复合物,以此促进羟基自由基的选择性,从而提升了难降解有机物的去除效果.
O3/MnO2;准好氧矿化垃圾床;紫外-可见光谱;三维荧光光谱;填埋场渗滤液
垃圾渗滤液是在垃圾填埋或焚烧处理过程产生,其含有大量的难降解的溶解性有机物(DOM),如憎水性腐殖质,约占DOM的40%~60%(质量分数)[1].根据在不同pH值下不同的溶解度,可将腐殖质分为富里酸(FA)、胡敏酸(HA)和胡敏素(HM);而非腐殖质物质,结构相对较简单,分子量较小,主要包括烃类、氨基酸、脂肪酸、糖类、酚酮类化合物和小分子有机酸等.基于“以废治废”的理念,准好氧矿化垃圾床处理渗滤液因其经济低耗而受到广泛关注[2-3].其采用渗滤液收集管和导气管末端与大气连通的方法,依靠矿化垃圾床内外温差产生的动力,形成“导气-进风”循环,继而通过床内并存的好氧、缺氧和厌氧区域加速体系中污染物的转化和降解[4].相比于传统垃圾渗滤液处理方法(如生物法、反渗透膜法及化学药剂等组合工艺)具有成本低、可操作性强的特点.研究发现,经准好氧矿化垃圾床对渗滤液氨氮去除效率可达98%,连续运行40周后,其垃圾渗滤液的色度和重金属均有较好的去除效果,尤其COD和总氮的平均去除率分别达到96.61%和95.46%[5],但渗滤液尾水依然存在COD和腐殖质浓度较高且可生化性较差的问题,因此还需对准好氧矿化垃圾床渗滤液尾水进行后续处理.
O3氧化技术因其处理高效,且无污泥产生,已经广泛运用于垃圾渗滤液中难降解有机物的深度氧化环节.为提高臭氧的利用率和氧化能力,O3/H2O2、O3/UV、O3/H2O2/UV等协同氧化技术发展较快,在一定条件下,对垃圾渗滤液的色度CN、腐殖质和COD的去除率可高达95%、78%、85%[6].工程应用上考虑到催化剂的可重复性以及分离成本,非均相催化剂相比于均相催化剂在催化臭氧氧化处理废水技术上有不可替代的作用,如课题组之前的研究认为O3/H2O2在用于废水处理上成本高达30~40元/kg COD[7];锰氧化物是一种环境友好型材料并且在土壤中广泛存在,因其成本低且易得,同时具有多种不同形式的变价氧化物的特点,使得锰基催化剂的研发及其催化O3技术成为了高级氧化领域的新趋势[7].相比之下,锰金属氧化物具有易分离、重复性强、经济效益高的特点.针对锰氧化物在高级氧化中的应用也有诸多报道,如竹湘峰等[8]对MnO-A催化剂中的MnO催化机理进行研究,表明MnO的引入可以明显增加催化剂表面羟基团的密度和活性,从而提高Mn催化剂的催化效率,这也证实了在非均相中锰氧化物存在多种催化路径;其次,Andreozzi等[9]也利用MnO2催化O3化草酸,表明反应机理是催化剂表面形成了“Mn-草酸”的复合物,加速了反应的进程,可以发现锰氧化物在不同废水处理过程中以不同的存在形式进行催化反应.由于众多的研究均是以模拟废水进行探讨,在实际过程中仍然缺乏技术支撑,并且利用准好氧矿化垃圾床联合O3氧化技术处理垃圾渗滤液的研究甚少,而针对老龄垃圾渗滤液中难降解有机物具有较强的化学稳定性,还能特异性猝灭羟基自由基的特点,尤其缺乏催化O3降解垃圾渗滤液中难降解有机物的效能和机理的相关报道.
鉴于此,构建了MnO2催化O3处理准好氧矿化垃圾床垃圾渗滤液尾水的方法,探究了单独O3和O3/MnO2体系处理尾水的区别,系统研究了O3投量、MnO2投量和初始pH值对有机物COD、腐殖质和色度CN三者去除率的影响;在此基础上,通过UV-Vis和3D-EEM技术分析了渗滤液尾水中DOM在体系降解过程中的分子结构变化;并运用XRD、SEM、EDS和XPS对MnO2表征了其在催化过程中的物相形态和价态的变化;最后,阐述MnO2在O3氧化降解过程中的催化机理及其在协同催化氧化体系中对有机物的降解规律,以期为O3/MnO2处理渗滤液尾水提供技术依据.
1 材料与方法
1.1 试验用水
垃圾渗滤液取自中国西部某大型传统厌氧型填埋场,运营时间为20a,其垃圾渗滤液呈深褐,水质偏碱性,pH值为7.86,COD为5680mg/L.实验所用废水经准好氧矿化垃圾床处理,准好氧矿化垃圾床装置已被多次报道[11],其对氨氮有十分优异的去除性能.渗滤液尾水呈浅棕色,pH值为7.2,COD为715mg/L,由于准好氧矿化垃圾床的微生物作用使得渗滤液尾水的可生化性极差,其水质特征如表1所示.

表1 准好氧矿化垃圾床预处理后水质情况
1.2 实验装置
实验装置如图1所示.主要由纯氧钢瓶、转子流量计(LZB-3WB)、臭氧发生器(KT-0Z-5G)、自制反应器(反应器尺寸:内径6cm,外径8cm,高度120cm,取样口距离底部2cm,容量规模约2.2L,为有机玻璃材质)、磁力搅拌器和2只KI 尾气吸收瓶组成.

图1 实验装置
1.3 仪器和试剂
主要仪器:分光光度计(Perkin-Elmer Lambda 950),微波快速 COD测定仪(APLMD-6型),三维荧光光谱仪(美国,HORIBA scientific,Aqualog-UV- 800C),分析天平(精度0.0001,BS 124S),酸度计(pHS-3C+).所用试剂无特殊说明的均为分析纯.
1.4 实验过程
将2.0L渗滤液尾水移至自制反应器中,通过稳流阀调节纯氧流量、改变臭氧发生器电流和臭氧浓度分析仪的校正,控制进气流量,该仪器对应臭氧转换量为氧气浓度0.1,0.2,0.3,0.4,0.5L/min,其O3产生量为9.798,18.92,32.16,43.68,52.65mg/min.分别以O3投加量、MnO2投加量和初始pH值为变量因素,加入预设量的MnO2,分别在不同的预设时间点,取样10mL并调节pH值到10.0,使用0.45 μm玻璃纤维膜过滤后,取样分析.
1.5 分析方法
依照《水和废水监测分析方法(第四版),采用重铬酸钾法测定COD;pH值采用玻璃电极法测定pH值(型号为成都方舟pHS-3C+);腐殖质的相对含量采用波长为254nm处的吸光度值表示(UV254);色度采用CN表示[12],如式(1):式中436、525、620分别表示在436,525和620nm波长时的吸光度值;

1.5.1 动力学拟合分析 COD、UV254和CN与反应时间的动力学拟合采用三次多项式(23)拟合,该多项式能较好地拟合两类体系中COD、UV254和CN与反应时间的关系(2均大于0.9900),采用软件Orgin 9.0进行拟合,取拟合式中b的值作为体系反应20min过程中的初始反应速率0[13];
1.5.2 紫外可见光谱分析 渗滤液尾水中难降解成分主要通过(型号Perkin-Elmer Lambda 950美国)紫外-可见分光光度计,扫描范围为220~600nm,扫描间隔为1nm.
1.5.3 三维荧光光谱分析 采用型号HORIBA scientific, Aqualog-UV-800C的三维荧光光谱仪测定.其中固定激发波长狭缝为5nm,扫描速度500nm/min,激发波长为239~550nm,发射波长250~650nm.
1.5.4 XRD分析 二氧化锰催化剂的结构通过型号为北京普析XD-2型的X 射线衍射仪(XRD)测定,其中,测是范围为5~90°,扫描速率为5°/min.
1.5.5 XPS分析 XPS采用型号为Thermo-VG250型X射线光电子能谱仪(XPS)测定.其中,单色Al~Ka (hv=1486.6eV),功率150W,500mm束斑.
1.5.6 SEM-EDS分析 形貌变化采用日本的JSM-5900LV扫描电子显微(SEM)进行分析,其中电压为20kV,分辨率为3.0nm.
2 结果与讨论
2.1 O3/MnO2与单独O3体系对渗滤液尾水中有机物去除的比较
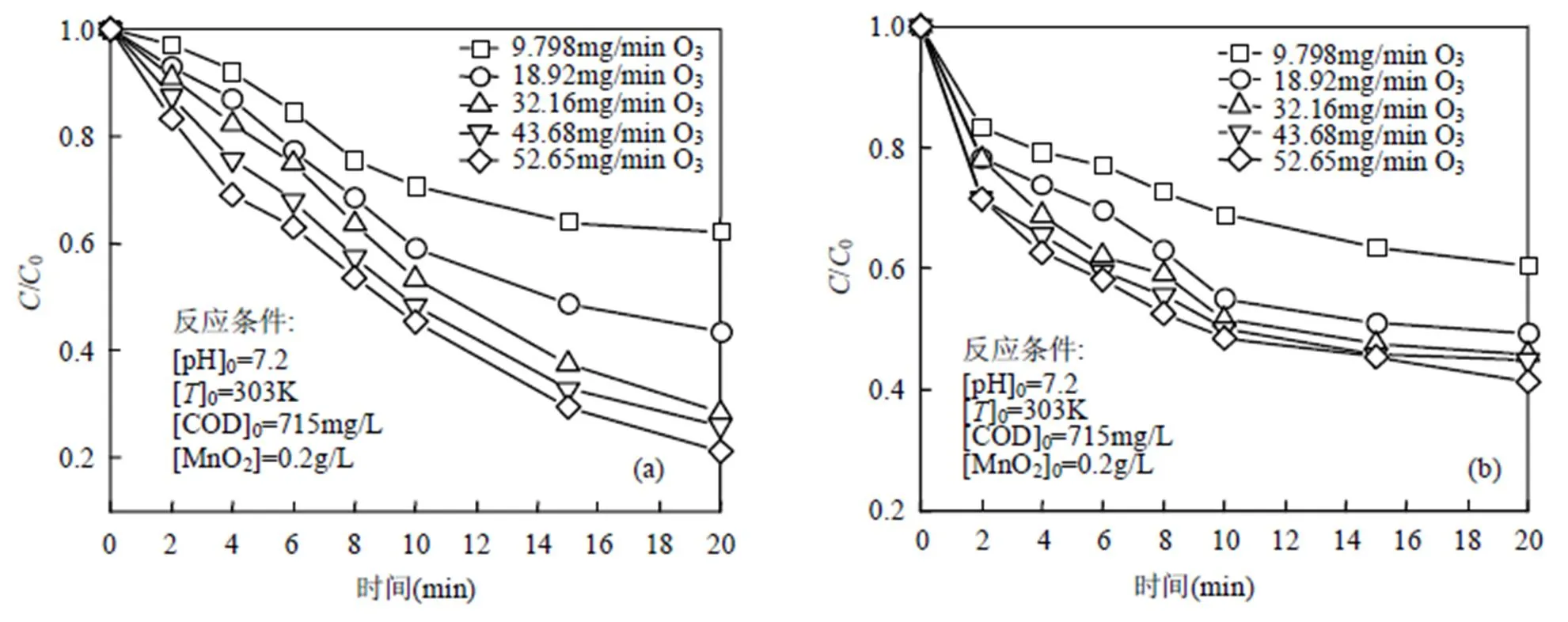
为验证MnO2催化特性,以COD和UV254作为考察指标,对单独O3体系与O3/MnO2体系进行对比研究,如图2单独O3与O3/MnO2氧化降解体系随时间变化情况所示.
由图2(a)知,当反应时间由2min延长至20min 时,O3/MnO2体系相比于单独O3体系对COD、UV254的去除率分别提升了24.66%、4.95%.另外,王春雨等[8]认为单独MnO2对废水中有机质的吸附量极少,并且其在氧气的作用下难以对大分子难降解有机物进行矿化作用.故MnO2对O3有较强的催化作用,能够大幅提升O3的氧化效率.同时,随着反应时间的延长,各实验组出水COD和UV254去除率均趋于平缓,这是因为O3在水中的溶解度很低,对污染物的去除率有一定的上限[14].王兵等[15]也认为O3在柱式反应器中, O3溶解饱和后,利用率受到溶解度的限制.
另一方面,通过污染物去除速率的动力学拟合结果可知,单独O3体系与O3/MnO2体系的相关系数2均较好(2>0.9900),COD和UV254初始反应速率分别由0.01、0.05大幅上升至0.05、0.08,这也表明O3/MnO2体系较单独O3能大幅提高准好氧矿化垃圾床渗滤液尾水中难降解有机污染物的去除效率,并且反应相对稳定.

图2 O3/MnO2体系对有机物去除率随时间变化情况
(a)对COD、UV254去除率的影响,(b)初始反应速率
2.2 影响因素分析
2.2.1 O3投加量 由图3可知,当O3投量从9.80mg/min增加至52.65mg/min,反应至20min时,体系对COD、UV254和色度CN的去除率分别从37.84%、39.68%和58.84%大幅上升至78.88%、58.74%和82.52%,动力学拟合也出现几乎相同的规律.相比之下,色度的去除率最大,这表明O3在降解体系中对带有显色基团的有机物有较好的去除效果,因为生色团中不饱和键发生了-1,3偶极环加成反应较为剧烈,之后腐殖质再逐渐被氧化分解为小分子有机产物,且腐殖质类废水在O3化过程中的脱色效果较为显著,这与周少奇等[16]研究结论一致.另一方面,随着反应进行到20min时,必然会出现O3化中间产物并逐渐积累,这就导致了O3/MnO2体系对3项污染物的去除率基本趋于稳定的状态;另外,袁建梅等[17]还认为过高的O3投量,会导致反应体系中大量生成的·OH相互发生复合反应,减弱O3/MnO2的催化氧化能力.因此,一定程度上增加O3的投量,可推动自由基链式反应,促进O3分解产生更多的羟基自由基,但过高的O3投量,会导致羟基自由基被消耗,降低去除速率.故将氧气流量设置为0.2L/min(此时O3投量为18.92mg/min)作为后续的反应条件.
(a)COD,(b)UV254,(c)色度CN,(d)对初始反应速率的影响

图4 二氧化锰投量对O3/MnO2体系的影响
(a)COD,(b)对UV254,(c)色度CN,(d)对初始反应速率的影响
2.2.2 MnO2投加量 MnO2投加量对O3/MnO2协同氧化体系氧化降解效率随时间变化的影响如图4所示.通过图4(a)可知,当MnO2投加量从0.1g/L增加至0.2g/L时,在反应至20min时,协同体系对COD、UV254、色度CN的去除率分别提升了13.28%、3.68%、10.64%;MnO2从0.2g/L增加至0.5g/L时,对3项污染物的去除率明显下降.之前的研究认为催化剂的浓度影响催化剂活性位,高浓度的催化剂可提供更多的活性位,但过多的 MnO2会在一定程度上催化O3分解成氧气,使O3浓度降低,进而产生的羟基自由基减少,导致降解率下降[18];另一方面,O3在MnO2的作用下发生一系列自由基链反应分解生成大量的·OH,由于·OH具有很高的氧化电位,当MnO2的用量超过一定值之后,会继续与OH·反应[8],从而降低了COD、UV254以及CN的去除率.从图4(d)污染物去除速率的动力学拟合结果可知, MnO2投加量在0.2g/L时,对COD、UV254和CN的去除速率最大,分别达到68.02%、50.74%和77.57%.
2.2.3 初始pH值 由图5可知,在初始 pH 值为3.0,反应时间为20min时, COD、UV254和色度CN的去除率分别达到65.00%、91.72%和62.01%.当初始 pH>3.0时,降解效果随初始pH值的升高而降低,这与郑可等[16]研究O3/H2O2均相O3氧化体系pH值越高,越利于羟基自由基的产生机制不同.渗滤液尾水中腐殖质的缓冲性能较强,在酸性条件下,其中COD降解生成小分子的酸类化合物和二氧化碳被释放,因此反应后pH值变化不明显;而在碱性环境下,矿化产物二氧化碳以及酸性产物的积累会使体系pH值降低速率加快.Andreozzi等[9]研究发现MnO2催化O3氧化有机物的本质是在催化剂表面形成了易被氧化的复合物,能够被羟基自由基氧化,且MnO2仅在酸性介质下表现出较强的催化性,因此随着pH值的升高,便不利于催化剂表面 “Mn氧化物-酸”复合物的形成,这会一定程度导致催化剂性能降低,从而使得对有机物的去除效果变差.由图5(d)污染物去除速率的动力学拟合结果可知,在酸性条件下,尤其是pH值为3时,三者的初始反应速率均较大.

图5 初始pH值对O3/MnO2体系的影响
(a)COD,(b)UV254,(c)色度CN,(d)对初始反应速率的影响
2.3 溶解性有机物在O3 /MnO2体系中的去除机理
2.3.1特征分子结构分析 紫外-可见光谱的吸收强度主要与含共轭双键有机物浓度有关,而紫外区的吸收强度与废水芳香性和复杂化程度有关[19],但是由于渗滤液尾水中DOM种类较多且浓度较高,故没有明显的吸收峰,但在紫外区具有较强的吸收强度.在O3/MnO2体系下不同反应时间的尾水的紫外可见光谱如图6所示.

图6 不同反应时间下渗滤液尾水的紫外-可见光谱
由图6可知,渗滤液尾水在紫外光区出现了较强的吸收,而在可见光区的吸收强度较低,说明该尾水中腐殖质具有较大的共振能量,芳香性也较高.随着反应时间的延长,出水的吸收强度在整个紫外光区都呈现了逐步下降的趋势[20].在特定波长下,紫外-可见(UV-Vis)的吸光度之比通常可以反映腐殖质的腐殖化程度、团聚及分子量的大小,其中254和280可以表征渗滤液尾水中有机物的芳香性和疏水性[21].由表2可知,随着反应时间的延长,其吸光度逐渐降低,显示有机物的芳香性和疏水性大幅度降低.而239~400(239~400nm处积分面积)表征苯环类化合物变化,随着反应的持续进行,其239~400不断减小,表明O3对尾水中苯环类化合物有较高的降解效果,而添加一定量的MnO2后,会提高O3对这类化合物的降解.一方面, MnO2的加入可以与溶解性有机物大分子形成复合物,使羟基自由基的选择性增强;另一方面, MnO2还能通过与体系中水结合形成水合MnO2,改变其催化剂的理化性质,促进羟基自由基的选择.故MnO2增大了羟基自由基的利用率,使得腐殖质被破坏,降低了其芳香性.

表2 MnO2/O3体系中渗滤液尾水特征吸光度变化
2.3.2 腐殖质结构分析 三维荧光光谱根据荧光物质的种类和位置划分为5个区域:芳香类蛋白-Ⅰ(x<250nm,m<330nm)、芳香类蛋白-Ⅱ(x< 250nm, 330nm
由图7可知,各区域均有荧光峰出现,各类荧光峰随反应时间的延长呈现出荧光值降低的趋势.由于紫外区类富里酸荧光主要由低分子量、高荧光效率的有机物引起,可见区类富里酸荧光则显示相对稳定、高分子量的芳香性类物质[23-24].首先,Flu2在反应2min时荧光强度大幅增加,其可能的原因是废水中存在一些固体有机物,在O3连续曝气的作用下,固体有机物会逐渐溶解导致了荧光强度的大幅提高;第二,Flu1和Flu2随着反应时间的延长,荧光强度逐渐降低,Flu1还发生了蓝移,说明难降解的紫外区类富里酸能较快地被O3/MnO2体系氧化降解.这表明腐殖质的分子结构能较快地得以破坏,降低分子的缩合度、芳香度和腐殖化程度[25].另外,由表3可知,原水中具有荧光效应的DOM相对较多,峰值较大,Ⅲ、Ⅴ区最大荧光峰位置出现在248/451nm、326/410nm处,分别代表类富里酸、类腐殖酸;经过O3/MnO2体系氧化降解20min后,出水的Ⅲ、Ⅴ区最大荧光峰位置出现在239/435nm、320/401nm处,峰强分别为409.59和259.28,去除率分别为76.67%和70.94%,且废水的可生化性也由进水的0.04大幅提升至0.35,表明O3/MnO2体系使渗滤液尾水的分子量和分子缩合度大幅度降低,有机物的腐殖度降低,可生化性得到提高.

图7 不同反应时间下渗滤液尾水的三维荧光

表3 渗滤液尾水的三维荧光特征峰及峰值变化
2.4 MnO2在体系中的催化机理
2.4.1 XRD分析 为探讨MnO2的催化性能,采用XRD表征反应前后MnO2的成分和形态变化,XRD扫描图谱如图8(a)所示.由图8(a)可知,对比其标准峰值线[26],可以发现反应前的5处特征衍射峰分别是21.46°、37.28°、42.68°、56.49°、67.18°,符合MnO2的特征峰值衍射线;反应后XRD图谱中5个特征峰均未发生移位,可以推断催化剂十分稳定,其成分形态并未发生改变.
2.4.2 SEM和EDS分析 为了揭示反应前后材料表面的形貌变化,扫描电镜以及EDS能谱图如图8(b)所示.由图8(b)可知,反应前(b1)的MnO2表面存在很多凹槽,催化剂表面不规则,且无其他杂质堆积;反应后(b2)的MnO2表面出现较大的块状物质,表面凹槽堆积大量一些细碎的物质.结合EDS图谱,选取[0~2keV]段,可知反应前后均富含O、Mn元素,反应后含有少量C、Ca元素.说明在大晶粒中锰氧化物占主要成分,即锰氧化物富集区;选取[2~4keV]段,可知反应后含S、Ca元素,且很微量,说明在晶界区域反应前无杂质,反应后为杂质富集区;选取[6~8keV]段,可知反应前后均仅含Mn元素,说明在小晶粒中无杂质进入主晶相[27].反应前球状颗粒分部均匀,这主要以锰氧化物为主,并且其富集区在大晶粒中,而反应后表面有块状物质填充,且比较光滑均匀,锰含量由25.45%降低到17.13%,这表明体系中含有的杂质锰化合物参与催化反应时,可能存在MnO2吸附有机物大分子形成复合物,增加羟基自由基的选择性,加速了氧化降解的速率,导致反应结束后表面吸附的少量有机物络合体,杂质锰化合物溶出.

图8 反应前后的MnO2的XRD及扫描电镜图
a. XRD图谱;b. SEM-EDS图谱
2.4.3 XPS分析 图9(a)为反应前后MnO2材料的XPS全扫描谱图.可以确定3个典型特征峰,分别为 C1s,O1s以及 Mn2p,由此可知复合材料主要由O、C和Mn 3种元素构成.分析反应前后的峰值图可以发现,反应后C1s和O1s的峰值有明显的下降,而Mn2p的峰值无明显变化.为了更加准确的分析出各元素的存在形态、种类及相对含量,利用XPSPEAK41软件对XPS全谱图进行分峰处理,由此得到MnO2材料的O1s 和Mn2p的分峰图谱如图9所示.
由Mn 2p分峰图9(b)看出,结合能在641.2,642.4和653.2eV分别对应Mn,Mn(Ⅱ)和Mn(Ⅳ).由表4可知,反应后Mn(Ⅱ)和Mn(Ⅳ)峰面积增大,代表含量均增加,而Mn0峰面积降低了541,表明Mn把大分子有机物还原成小分子有机物,又由于Mn(Ⅱ)峰面积积累量最大达到2706.73,推测Mn主要转化为了Mn(Ⅱ).而通过表4可以发现,反应后Mn(Ⅳ)的占比最高达到37.49%,因此可认为Mn(Ⅳ)占主导地位,由此可推断出MnO2作为O3/MnO2体系催化剂催化性质稳定,符合其催化机理主要为MnO2催化O3产生羟基自由对有机物进行氧化降解机制的推断[29-32].

a.全扫描图谱; b.O1s分峰图谱; c.Mn2p分峰图谱

表4 反应前后MnO2催化剂XPS的分峰拟合结果
由O1s分峰图9(c)看出,结合能在528.9,530.9和532.5eV分别对应O2-,OH-和H2O,结合表4可以发现反应前后OH-峰面积仅变化128.62,说明反应后溶液酸碱性变化不大,酸性条件下几乎不改变,这与单因素分析测定的反应后pH值无明显变化结论相符,这一现象说明O3/MnO2体系在有机物的去除过程中,主要由O3产生的羟基自由基进行无选择的氧化作用,对有机物进行去除.而O2-的含量提高可能是由于MnO2体系中含有少量的杂质氧化物在酸性条件下,进行溶出得以去除[28],这与EDS表征结果一致.
2.5 O3/MnO2体系对难降解有机物的机理探讨
由于MnO2本身具有很稳定的催化性质,对O3/MnO2体系有机物的去除有较为明显的提升.而在催化过程中,其反应机理主要利用了×OH强的氧化性,由于×OH的无选择性,因此可将多数有机物氧化. MnO2催化剂的加入使得反应体系中产生了更多的活性物种且主要是羟基自由基,同时MnO2自身也通过改善其表面理化性质参与降解反应.
2.5.1 体系自由基氧化机制 实验表明低浓度的MnO2投量可以促使O3产生羟基自由基,因此从MnO2投量的影响变化中可推测出过量投加MnO2,在一定程度上会降低对有机物的去除率,这与Ma Jun等[33]研究认为新生态的MnO2催化氧化降解阿特拉津遵循自由基反应机理的结论相符.因此,在O3/MnO2体系中起主要作用的是O3及其体系产生的以羟基自由基为主的活性物种,其降解式如(2)、(3).

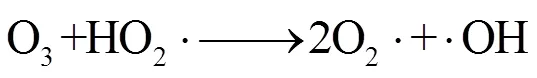
2.5.2 无机-有机复合物的氧化机制 在pH值的降解变化关系中可以推测MnO2催化O3氧化化有机物的本质是在催化剂表面形成了易被氧化的复合物,其反应机理主要是在MnO2表面形成了“锰氧化物-有机物分子”的复合物,使得羟基自由基能够更容易的降解此类复合物.如式(4)所示,使缺乏选择性的羟基自由基更容易与之结合,达到氧化降解的目的,这也与Andreozzi等[9]用MnO2非均相催化O3氧化草酸时的结论相符.表面羟基或 O3吸附于水合MnO2表面,作为连接污染物和催化剂的桥梁,加速污染物与催化剂的相互接触,促进整个降解反应.

综上, MnO2、MnO2复合物以及水合MnO2催化O3的复合催化机制使非均相环境介质转变为水溶液、催化剂的表面和催化剂-溶液界面等 3 种环境[34].故MnO2催化O3氧化处理渗滤液尾水的过程中,由于MnO2催化O3产生羟基自由基以及有机物复合促进羟基自由基的催化机制使得准好氧矿化垃圾床渗滤液尾水中难降解有机物得以快速去除,且可生化性大幅提高.
3 结论
3.1 O3/MnO2体系较单独O3对渗滤液尾水中难降解有机物有显著的提升效果,当MnO2投加量为0.2g/L,O3进气量为18.92mg/min,初始 pH值为3,反应时间为20min时,体系对COD、UV254、色度CN的去除效果较好,其去除率分别为63.79%、57.68%、90.79%.
3.2 渗滤液尾水中特征大分子有机物腐殖质在O3/MnO2体系中被有效降解,可生化得到显著提升,为后续的生化反应提供了可能.
3.3 MnO2的表征显示,反应前后MnO2的形态未出现明显变化, Mn(Ⅳ)在样品中占主导地位,其催化机理为MnO2催化O3产生羟基自由以及有机物复合促进羟基自由基的催化机制.
[1] 熊建英,郑 正.垃圾填埋场渗滤液溶解性有机质特性及其去除技术综述[J]. 环境化学, 2015,34(1):44-53.
[2] 方 芳,刘国强,郭劲松,等.渗滤液中DOM的表征及特性研究[J]. 环境科学, 2009,30(3):834-839.
[3] 李鸿江,赵由才,柴晓利,等.矿化垃圾反应床处理垃圾渗滤液出水中的水溶性有机物[J]. 环境污染与防治, 2008,30(11):4-8.
[4] 何水晶.连续进水条件下准好氧矿化垃圾床处理渗滤液的试验研究[D]. 成都:西南交通大学, 2008.
[5] Han Z Y, Liu D, Li Q B. A removal mechanism for organics and nitrogen in treating leachate using a semi-aerobic aged refuse biofilter [J]. Journal of Environmental Management, 2013,114(2):336-342.
[6] Katsoyiannis A I., Canonica S, Ursvon G. Efficiency and energy requirements for the transformation of organic micropollutants by ozone, O3/H2O2and UV/H2O2[J]. Water Research, 2011,45:3811-3822.
[7] 王春雨,侯永江,刘 璇,等.不同晶型二氧化锰催化臭氧化降解亚甲基蓝废水[J]. 环境工程学报, 2017,11(2):908-914.
[8] 竹湘锋.有机废水的催化臭氧氧化研究[D]. 杭州:浙江大学, 2005.
[9] Andreozzi R,Insola A,The kinetic of Mn(Ⅱ)/Catalyzed Ozonation of Oxalic Acid in Aqueous Solution [J]. Water Research, 1992,17:917-921.
[10] 陈炜鸣,张爱平,李 民,等.O3/H2O2降解垃圾渗滤液浓缩液的氧化特性及光谱解析[J]. 中国环境科学, 2017,37(6):2160-2172.
[11] 蒋国斌,寇 月,李启彬,等.导气管管径对准好氧生物反应器填埋场碳氮污染物转化的影响[J]. 环境科学学报, 2017,37(4):1427-1435.
[12] 张爱平,李 民,陈炜鸣,等.臭氧降解村镇垃圾中转站渗滤液中有机污染物的试验研究[J]. 环境科学学报, 2017,37(2):694-702.
[13] 郑 可,周少奇,沙 爽,等.臭氧氧化反渗透浓缩垃圾渗滤液动力学[J]. 环境科学, 2011,32(10):2966-2970.
[14] Qi C, Liu X, Lin C, et al. Activation of peroxymonosulfate by microwave irradiation for degradation of organic contaminants [J]. Chemical Engineering Journal, 2017,315:201-209.
[15] 王 兵,税奕瑜,刘璞真,等.臭氧在柱式反应器中的强化传质过程[J]. 环境工程学报, 2017,11(2):760-768.
[16] 郑 可.臭氧氧化法处理反渗透浓缩垃圾渗滤液[D]. 广州:华南理工大学, 2012.
[17] 袁建梅,杨德敏.非均相催化臭氧化深度处理钻井废水的效能研究[J]. 工业水处理, 2014,34(8):36-40.
[18] 黄行康.二氧化锰的制备、结构表征及其电化学性能[D]. 厦门:厦门大学, 2007.
[19] Tizaoui C, Bouselmi L, Mansouri L, et al. Landfill leachate treatment with ozone and ozone/hydrogen peroxide systems [J]. Journal of Hazardous Materials, 2007,140(1/2):316-324.
[20] Kang K H, Shin H S, Park H. Characterization of humic substances present in landfill leachates with different landfill ages and its implications [J]. Water Research, 2002,36(16):4023-4032.
[21] 崔东宇,何小松,席北斗,等.堆肥腐熟前后胡敏酸与富里酸的还原容量比较[J]. 中国环境科学, 2015,35(7):2087-2094.
[22] Chen W, Westerhof P, Leenheer J A, et al.Fluorescence excitation-emission matrix regional integration to quantify spectra for dissolved organic matter [J]. Environmental Science & Technology, 2003,37(24):5701-5710.
[23] 贾陈忠,王焰新,张彩香,等.垃圾渗滤液中溶解性有机物组分的三维荧光特性[J]. 光谱学与光谱分析, 2012,32(6):1575-1579.
[24] He X S, Xi B D, Gao R T, et al. Insight into the composition and degradation potential of dissolved organic matter with different hydrophobicity in landfill leachates [J]. Chemosphere, 2016,144:75-80.
[25] Andy B, Michael C. Fluorescence of leachates from three contrasting landfills [J]. Water Research, 2004,38:2605-2613.
[26] Guo J H, Dong Y T, Zhang D Y. Dissolved Silica Adsorbed onto MnO2in Catalyzing the Decomposition of H2O2: Molecular Structural Rearrangement Revealed by FTIR, SEM-EDX and XRD [J]. Agricultural Science & Technology, 2017,18(10):1912-1915.
[27] Wang Y, Sun H, Ang H M, et al. Facile synthesis of hierarchically structured magnetic MnO2/Zn Fe2O4hybrid materials and their performance in heterogeneous activation of peroxy monosul fate [J]. ACS Appl Mater Interfaces, 2014,6(22):19914-19923.
[28] Jiang J, Zhang X, Sun P, et al. ZnO/BiOI Heterostructures: Photo induced Charge-Transfer Property and Enhanced Visible-Light Photocatalytic Activity [J]. Journal of Physical Chemistry C, 2011, 115(42):20555-20564.
[29] Xiao W, Wang D, Lou X W. Shape-Controlled Synthesis of MnO2Nanostructures with Enhanced Electrocatalytic Activity for Oxygen Reduction [J]. Journal of Physical Chemistry C, 2009,114(3): 1430-1434.
[30] Du Y, Ma W, Liu P, et al. Magnetic Co Fe2O4nanoparticles supported on titanate nanotubes (Co Fe2O4/TNTs) as a novel hetero geneous catalyst for peroxymonosulfate activation and degradation of organic pollutants [J]. Journal of Hazardous Materials, 2016,308:58-66.
[31] Kang M, Park E D, Kim J M, et al. Simultaneous removal of particulates and NO by the catalytic bag filter containing MnOx catalysts [J]. Korean Journal of Chemical Engineering, 2009,26(1):86-89.
[32] Yao Y, Cai Y, Wu G, et al. Sulfate radicals induced from peroxymonosulfate by cobalt manganese oxides (Co(x)Mn(3-x)O4) for Fenton-Like reaction in water. [J]. Journal of Hazardous Materials, 2015,296:128.
[33] Ma J, Graham N J D. Preliminary Investigation of Manganese- Catalyzed Ozonation for the Destruction of Atrazine[J]. Ozone Science & Engineering, 1997,19(3):227-240.
[34] Meng Y, Song W, Huang H, et al. Structure-property relationship of bifunctional MnO2nanostructures: highly efficient, ultra-stable electrochemical water oxidation and oxygen reduction reaction catalysts identified in alkaline media [J]. Journal of the American Chemical Society, 2014,136(32):11452-11464.
Removal of refractory organics from SAARB treated landfill leachate by O3/MnO2process.
DENG Yu-nan, CHEN Wei-ming, LUO Zi-yin, CUI Yu-qi, LI Qi-bin*
(Geosciences and Environmental Engineering of Southwest Jiaotong University, Chengdu 611765, China)., 2018,38(11):4130~4140
A large number of refractory organics residual in semi-aerobic aged refuse biofilter (SAARB) leachate, ozone/manganese-dioxide (O3/MnO2) process was used to catalytically decompose organics from SAARB treated landfill leachate. Effects of ozone dosage, MnO2dosage and initial pH on the removal of organic substances and reaction kinetics were investigated. UV-Vis and 3D-EEM tests were applied to investigate the transformation mechanism of recalcitrant organics in O3/MnO2process. In addition, the phase change of MnO2before and after reaction and its catalytic mechanism were investigated by SEM, EDS, XRD and XPS. Results showed that when the ozone dosage of 18.92mg/min, initial pH of 3 and reaction time of 20min, compared to the ozone alone treatment, the O3/MnO2peroxide treatment was significantly improved with 2mg/L MnO2addition. The removal efficiencies of COD, UV254, and CN by 24.66%, 4.95%, and 12.57%, respectively, and the chromophore was most easily attacked by ozone. UV-Vis spectra and 3D-EEM spectrum were both illustrated that O3/MnO2process can significantly decrease the aromaticity degree, molecular weight and condensation degree of organic substances in wastewater, which degradation efficiency of benzene ring compounds was improved significantly and also greatly proved biodegradability of leachate effluent. Meanwhile, After O3/MnO2process, MnO2has not shown the change of the peak value of the new valence state, and Mn(Ⅳ) played a dominate role in the catalytic process. In order to promote the selectivity of hydroxyl radical and the catalytic performance, indicating that the mechanism of O3/MnO2process was MnO2catalyzed O3to generate hydroxyl radicals and transformed into hydrated manganese dioxide, which changed the physicochemical properties of the catalyst surface.
O3/MnO2;semi-aerobic aged refuse biofilter;UV-Vis;3D-EEM;landfill leachate
X703.5
A
1000-6923(2018)11-4130-11
邓禺南(1993-),男,四川成都人,西南交通大学环境工程专业硕士研究生,主要从事难降解有机废水的处理技术研究.
2018-04-13
四川省高校特种废水处理重点实验室开放课题(SWWT2015-4)
* 责任作者, 教授, liqb@home.swjtu.edu.cn
