硝酸异丙酯水解反应的量子化学计算
2018-11-12陈永康安振涛陈明华
陈永康,安振涛,陈明华,张 力
(1.陆军工程大学石家庄校区,河北 石家庄 050003;2. 特种勤务研究所,河北 石家庄 050003)
引 言
硝酸异丙酯是一种硝酸酯类的含能材料,不仅可以作为战斗部装药,也可用于火箭推进剂。除燃爆特性以外,它还具有强氧化性、强挥发性和毒性,废弃的硝酸异丙酯需要及时进行处理,否则将造成严重的安全隐患。而当前还没有针对硝酸异丙酯的安全快速去能处理方法,通常是采用挥发、烧毁、炸毁等方式处理,存在环境污染严重、危险性大、资源浪费等问题,不符合当前“绿色环保、安全高效”的要求,因此寻找安全绿色的硝酸异丙酯处理方法是亟待解决的问题。
根据硝酸异丙酯属于酯类的特点,对其进行水解是一种相对有效的处理方法。目前,关于硝酸异丙酯性质和水解反应机理已有一定的理论研究[1-6]。曾秀琳等[7]采用密度泛函方法计算了其前线轨道能并分析了其对热稳定性的影响;True等[8]通过实验探索了硝酸异丙酯的构型;Thornton[9]根据经典力学对其水解机理做出了理论推断;贡雪东等[10]运用SCF-MOAM1方法确定了硝酸异丙酯的几何构型,并通过AM1方法得到了气相状态下硝酸异丙酯的SN2碱性水解位能曲线;刘志勇等[11]分别用AM1和B3LYP/6-31+G(d)计算了气相状态下硝酸异丙酯的热分解初始反应机理和水解反应机理。
本研究在此基础上,采用更高精度的计算方法,考虑其溶剂化效应及pH值的影响,计算了反应动力学,对硝酸异丙酯的水解机理进行研究。
1 计算方法
所有的电子结构和能量计算均由Guassain 09程序包完成。采用从头算二阶多体微扰理论(second order Mφller-Pleset perturbation theory, MP2) 6-311+g(d,p)基组优化各反应中反应物、产物和过渡态的几何结构并计算振动频率,MP2方法要求在Hartree-Fock计算之后接着进行Mφller-Pleset关联能校正计算,对MP2截断到二阶。对反应过渡态进行内禀反应坐标(IRC)计算以对过渡态加以确认。反应体系为带电体系,对体系进行闭壳层计算。采用极化连续介质模型(Polarizable Continuum Model, PCM)研究了反应的溶剂化效应。在MP2/6-311+g(d,p)水平下计算得到标准态和溶剂化条件下的反应能级图。为确定pH值对水解反应的影响,在MP2/6-311+g(d,p)水平计算得到不同pH值时反应物、产物、过渡态及络合物的能量。
反应的动力学计算由Polyrate 2015程序完成。在CCSD(T)/6-311++g(3df,2p)水平下,通过经典过渡态理论 (TST),正则变分过渡态理论(CVT),零曲率隧道效应校正的正则变分过渡态理论 (CVT/ZCT)和小曲率隧道效应校正的正则变分过渡态理论 (CVT/SCT) 对反应速率常数进行计算。
2 结果与讨论
2.1 反应路径
对亲核取代SN2反应和β-H消除Eβ反应这两种反应路径进行了计算比较,两种反应路径如图1所示。亲核取代反应(以下称为R1)的产物为异丙醇和硝酸根离子,β-H消除反应(以下称为R2)的产物为丙烯,硝酸根离子以及水。
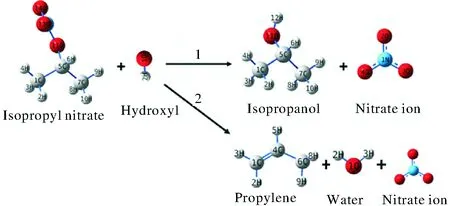
图1 硝酸异丙酯碱性水解反应Fig.1 The alkaline hydrolysis reaction of isopropyl nitrate
结合图1,根据硝酸异丙酯的几何结构可以发现,氢氧根离子进攻中间碳原子仅有一种路径,而氢氧根进攻末端氢原子时,由于氢原子不具有对称性,因此有3种路径。即本研究中的硝酸异丙酯碱性水解过程共包括4条反应路径:
Pathway 1:氢氧根离子从硝酸异丙酯背面进攻中间碳原子,发生SN2亲核取代反应,反应产物为异丙醇和硝酸根离子。
Pathway 2:氢氧根离子进攻硝酸异丙酯末端碳上的9号氢原子,生成丙烯、水、硝酸根离子。
Pathway 3:氢氧根离子进攻硝酸异丙酯末端碳上的10号氢原子,生成丙烯、水、硝酸根离子。
Pathway 4:氢氧根离子进攻硝酸异丙酯末端碳上的8号氢原子,生成丙烯、水、硝酸根离子。
2.2 几何构型
根据以上4种反应路径,确定各路径的过渡态,然后在MP2/6-311+g(d,p)水平下优化各路径的反应物、过渡态及产物的几何结构,如图2所示。

图2 所有反应物和产物的几何结构Fig.2 Geometries of all reactants and products
对于硝酸异丙酯的构型,研究表明,O—NO2具有平面结构,C—O—NO2基本共面,硝酸酯基本不可能绕O—NO2旋转。同时也普遍认为硝酸异丙酯为反式构型,但与硝酸正丙酯不同的是,由于支链的影响,硝酸异丙酯中的C—O—NO2并不像硝酸正丙酯一样完全共面,其C5O11N12O14二面角约为178°。
结合驻点结构,对于两种反应,在R1中,氢氧根从背面进攻中间的碳原子C5,氢氧根的O逐渐靠近C5,首先形成络合物1, C5—O11键伸长,形成过渡态;然后氢氧根失电子与C5结合,C5—O11键断裂,O11得电子,形成络合物2;最后,络合物2分解生成硝酸根离子和异丙醇。在R2中,氢氧根离子进攻硝酸异丙酯末端碳上的氢原子,首先形成络合物1,氢氧根的O逐渐靠近末端氢原子,C5—O11键伸长,形成过渡态;而后末端碳原子的C-H键断裂,失去的氢原子与氢氧根结合,末端碳原子与C5形成碳碳双键,C5—O11键断裂,O11得电子,形成络合物2;最终络合物2分解生成丙烯、水和硝酸根离子。
2.2 前线轨道能和频率计算
在同水平下计算得到3种物质的前线轨道能,最低未占分子轨道能(ELUMO)和最高已占分子轨道能(EHOMO),并计算能级差(ΔE)。其中ΔE=ELUMO-EHOMO,结果见表1。一般认为,能级差越小,电子越容易从最高已占分子轨道向最低未占分子轨道跃迁,分子就越不稳定。

表1 主要反应物和产物的前线轨道能
根据表1中的结果,反应物硝酸异丙酯的能级差最小,两种产物中异丙醇的能级差高于丙烯,一定程度上说明异丙醇比丙烯更加稳定。曾秀琳等[7]采用密度泛函方法B3P86/6-31G*计算得到硝酸异丙酯的能级差(ΔE)为0.2508hartree,比本研究MP2方法的计算结果小,其原因可能是B3P86方法计算了更多的电子相关、电子排斥效应,因此所得结果更小。
在同水平下计算各反应物、产物及过渡态的振动频率,反应物和产物的频率均为正值。异丙醇的O—H伸缩振动频率的计算值和实验值分别为3879和3819cm-1;氢氧根振动频率的计算值和实验值分别为3269和3309cm-1,计算结果与相关的实验值较为吻合。过渡态有且仅有一个虚频。通道1的过渡态TS1的虚频为600i,通道2~4的过渡态TS2、TS3、TS4的虚频相互接近,分别为1101i、1154i、1107i。
2.3 溶剂化效应
水解反应实际是在溶液中进行的,分子和过渡态在气态和在溶液中的性质会存在较大区别,溶剂化效应会对分子能量和反应能垒等造成影响[12-16]。而目前尚未见关于该反应溶剂化效应的研究报道,因此本研究采用极化连续介质模型(PCM)对反应的溶剂化效应进行研究。
在MP2/6-311+g(d,p)水平下对各反应物和产物在标准态和溶剂化条件下的能量同时进行计算,得到所有反应物和产物在标准态和溶剂化条件下的结构能量(E)和零点能校正(ZPE),结果见表2。计算得到两种反应在标准态和溶剂化条件下的反应焓变(ΔH),结果见表3。

表2 所有反应物和产物的热力学数据

表3 R1与R2的反应焓变
由表3可知,溶剂化条件下两种反应的反应焓比标准态高约80kJ/mol,另外,两种条件下R2的反应焓都要高于R1。文献[11]计算得到的标准态下R1的理论值与本研究计算结果接近。
图3为MP2/6-311+g(d,p)水平下计算得到的经零点能校正后标准态和溶剂化条件下的反应能级示意图,各驻点能量按图例顺序列出。

图3 标准态和溶剂化条件下的反应能级示意图Fig.3 Schematic diagram of reaction level under standard state and solvation conditions
由图3可知,在标准态下,反应物首先生成中间体IM1,IM1经由过渡态生成IM2,这一步Pathway1至Pathway4的能量差分别为55.86、45.61、27.91、33.85kJ/mol,最后IM2越过能垒生成产物。溶剂化条件下反应的能垒顺序与标准态下基本一致。但不同的是在溶剂化条件下,反应势能曲线的能量远低于标准态下反应势能曲线的能量。
在硝酸异丙酯水解的所有反应路径当中,Pathway 3(s)的过渡态能垒最低。标准态下,Pathway 2过渡态能垒最高,Pathway 3过渡态能垒最低;溶剂化条件下,Pathway 1(s)过渡态能垒最高,为82.92kJ/mol,Pathway 3(s)过渡态能垒最低,为60.04kJ/mol,这意味着在低温下,反应易朝着Pathway 3(s)的方向进行。然而对比产物能量可知,Pathway 1(s)的产物能量最低,说明在高温下,当能量足够越过反应能垒时,反应倾向于Pathway 1(s)方向,生成稳定的异丙醇和水。
贡雪东等[10]通过半经验AM1方法求得气相条件下R1的活化能为75.73kJ/mol;刘志勇等[11]用B3LYP/6-31+G(d)计算了气相状态下R1的活化能为52.51kJ/mol。本研究计算得到的R1标准态下的过渡态能垒比文献值高,意味着水解反应在实际情况下可能不容易进行。
2.4 pH值对水解反应的影响
为确定溶液pH值对水解反应的影响,在MP2/6-311+g(d,p)水平计算得到不同pH值时反应物、产物、过渡态及络合物的能量,如表4所示。从表4中可以看出,当pH值为8~12时,各驻点能量基本不变,当pH值继续变大达到13、14时,驻点能量略有升高,总之,pH值的变化对反应物、络合物1、过渡态、络合物2、以及产物的能量影响不大,该计算结果表明,pH值不影响反应路径顺序。其原因可能是pH值的变化主要影响水解反应的反应速率,对能量的影响较小。

表4 不同pH值下所有反应物(R)、过渡态(TS)、络合物(IM1和IM2)和产物(P)的能量
2.5 动力学计算
在CCSD(T)/6-311++g(3df,2p)水平,气态条件下,通过TST、CVT、CVT/ZCT和CVT/SCT理论方法[17-19]对273~373K温度范围内R1的反应速率常数进行计算,得到的反应势能面如图4所示,4种方法计算出的反应速率常数随1000/T的变化曲线见图5。
由图4和图5可知,随着温度的升高,反应速率常数逐渐增大。但总体而言,计算得到的该反应的速率常数较小,表明在实际情况下,反应速率较慢。同时,MP2方法计算速率常数值比实际情况偏小,可能会导致计算结果较小。
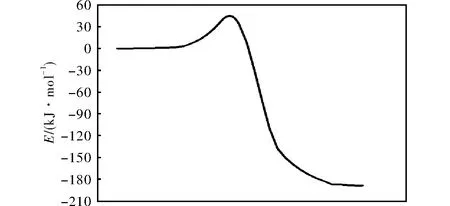
图4 R1的反应势能面Fig.4 Potential energy surface of R1
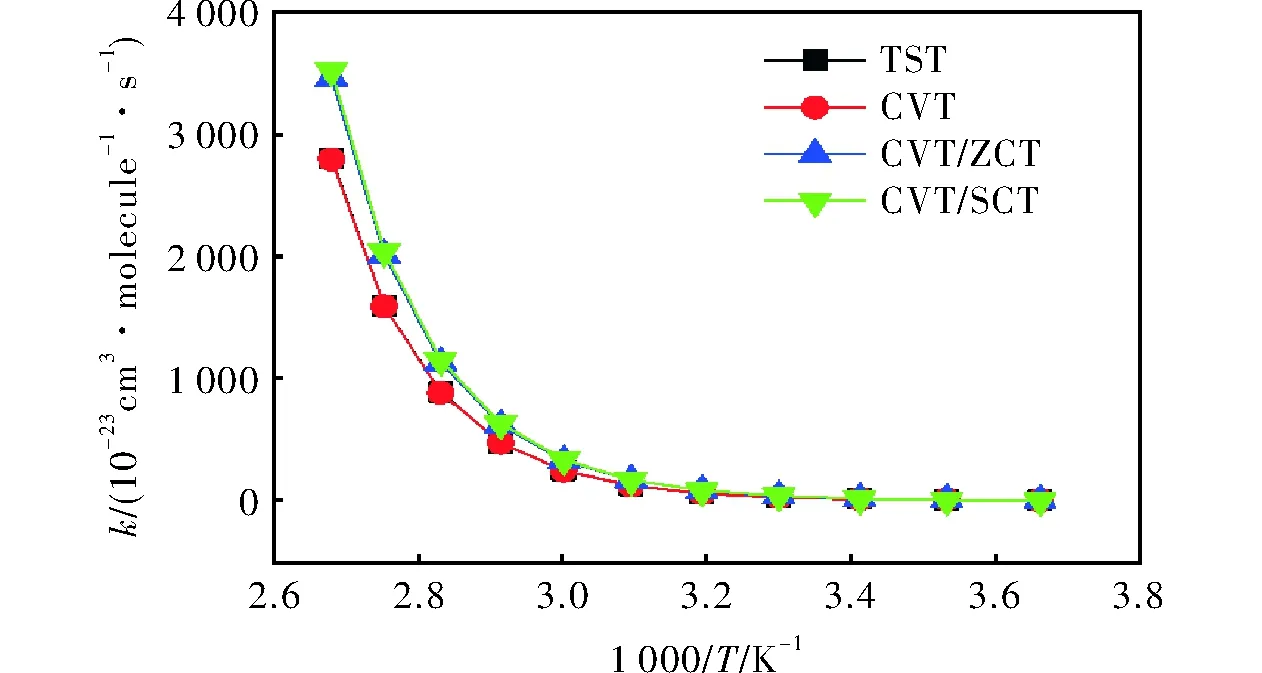
图5 R1的反应速率常数随1000/T的变化曲线Fig.5 Reaction rate constants vs. 1000/T curves of R1
另外,TST和CVT 方法的计算值接近,这意味着整个过程中的变分效应很小,所得结果准确。TST、CVT方法的速率常数计算结果与CVT/ZCT、CVT/SCT方法相比,在低温处基本一致,在高温处略有偏差,表明在高温处的隧道效应对计算结果有一定影响。
在273~373K范围内,假定活化能不随温度发生改变,根据Polyrate的计算数据,对lnk和1/T进行线性拟合,得到反应速率常数k的Arrhenius表达式为:k=1.18×10-23exp(-7357/T),k单位为cm3/(molecule·s)。同时,对原始数据进行非线性拟合,得到三参数的Arrhenius表达式为:k=9.14×10-22(T-0.47)exp(-9949/T),k的单位为cm3/(molecule·s)。目前,有关该反应速率常数的研究较少,因此此计算结果可为今后进一步研究提供参考。
3 结 论
(1)采用从头算方法和基组优化反应路径中各驻点的几何结构并计算前线轨道能和振动频率,采用极化连续介质模型研究了反应的溶剂化效应,得到标准态和溶剂化效应下的反应势能面,对比发现在高温下反应更倾向于生成稳定的异丙醇和水,其过渡态能垒为82.92kJ/mol。
(2)研究了pH值变化对水解反应的影响,发现pH值的变化对驻点能量以及反应路径顺序影响不大。利用TST、CVT、CVT/ZCT和CVT/SCT方法得到SN2反应速率常数k的Arrhenius表达式为:k=1.18×10-23exp(-7357/T),计算出的反应速率常数相对较小,说明反应速率可能较慢。
致谢:感谢华南师范大学提供Gaussain 09和Polyrate 2015程序包;同时感谢王朝阳老师在研究过程中给予的指导和帮助。
