基于排序融合模型的紫癜性肾炎患者中差异表达变量的筛选研究*
2018-11-05哈尔滨医科大学公共卫生学院卫生统计学教研室150081
哈尔滨医科大学公共卫生学院卫生统计学教研室(150081)
高 兵 刘美娜 谢 彪 王玉鹏 孙 琳 张秋菊△
【提 要】 目的 对紫癜性肾炎和过敏性紫癜两类患者中差异表达的代谢产物进行筛选。方法 利用排序融合的思路,将t检验、Wilcoxon秩和检验、偏最小二乘、及随机森林等四种方法用于组间差异表达分析,对其所获得的四个变量重要性排序进行融合,获得综合的、单一的变量排序(排序融合模型);利用交叉验证获得最优模型,并进行差异变量的筛选;通过模拟实验评价排序融合模型变量筛选的能力并与least absolute shrinkage and selection operator(LASSO)进行比较。最后,将其用于紫癜性肾炎与过敏性紫癜患者间的代谢物差异分析。结果 模拟实验结果显示:(1)当观测数和差异变量数较小时,排序融合模型的AUC的平均值大于LASSO;(2)当观测数和差异变量数较大时排序融合模型的AUC的平均值与LASSO相近;3)无论参数如何设置排序融合模型所筛选的差异变量数基本均少于LASSO。实例分析结果显示:应用排序融合模型获得紫癜性肾炎和过敏性紫癜患者中存在12个差异表达的代谢产物,其AUC值达到其最大值0.96。结论 相比于LASSO,排序融合模型在筛选变量时更具可靠性和准确性,可为代谢组学数据的差异表达分析提供新的分析思路和方法。
近年来许多的变量筛选方法被提出来,包括单变量方法和多变量方法。单变量方法筛选变量时不基于任何模型,如t检验和Wilcoxon秩和检验可用于检验两组不同人群之间某一连续性变量是否有差异,但分析高维组学数据时,因未考虑变量之间的相关性,而导致犯第I类错误的概率增加[1]。多变量分析方法筛选变量往往基于特定模型,根据所属类型又分为模型拟合与模型预测。前者代表如偏最小二乘(partial least square,PLS),属于有监督方法,结合了主成分(principal component analysis,PCA)、多元线性回归和典型相关分析的特点,优势是突出组间差异,但有时会出现模型过拟合[2-3]。后者代表如随机森林(random forest,Rforest)利用bootstrap重抽样法抽取样本并进行决策树建模,具有避免过拟合、抗噪的优势,但模型会存在一定限度内的泛化误差[4]。
在高维组学数据分析中,无论单变量还是多变量方法,对于同一组数据若采用不同的分析方法,所筛选出的差异变量通常不一致,并且各差异变量在重要性排序中所在位置也不尽相同。为此本研究考虑各个方法对变量筛选的影响,利用排序融合思想建立排序融合模型,整合不同变量筛选方法的排序信息形成融合序列,并对紫癜性肾炎患者中差异表达变量进行筛选。
模型介绍
1.排序融合模型
排序融合思想[5]首先定义一个函数,将不同变量筛选方法的变量排序融合问题转化成求最优解,形式如下:
Li是第i个变量筛选方法的变量排序列,wi为第i个变量筛选方法的变量排序列的权重,d是一个距离函数,δ是一个与Li长度相同的变量排序列。
排序融合致力于得到与所有变量排序都相似的序列,因此,最优化问题转化为求与每一个变量排序都尽可能相似的排序。从公式看,就是求使δ与所有的Li距离最近的最优δ*:
距离函数选用Spearman简捷距离函数[6]。Spearman简捷距离函数擅于处理只有排序信息的变量序列。δ与所有Li的Spearman简捷距离函数定义如下:
rLi(A)表示序列Li中物质A的排序(1代表排第一),如果物质A排在前k,rLi(A)赋值为k,没有排在前k的赋值为k+1;rδ(A)赋值方式与rLi(A)相同。Spearman简捷距离最大为k(k+1),此时两个序列完全不一样。本研究中生成融合序列的四种变量筛选方法包括t检验、Wilcoxon 秩和检验、PLS和Rforest。
2.利用排序融合模型实现变量筛选
首先求出最优的融合变量排序,然后利用这个序列进行变量筛选。具体的做法是:通过logistic模型的2折交叉验证,将融合序列中的变量按照排序加到logistic模型中,直至模型的分类准确率和AUC值达到最大值,此时加入模型中的变量即为筛选的差异变量[7-8]。
模拟实验
1.模拟实验条件设置及模拟数据产生
在代谢组学数据中,变量通常呈偏态分布,并且包含极端值,为此设置病例组和对照差异变量分别服从X~E(0.1)和X~E(0.19)的指数分布。随机产生1000个多元标准正态分布噪声变量,将噪声分为20组,每50个噪声之间的相关系数设为0.95。分别模拟差异变量为5、10、30、50和样本量为20、40、60、100时,比较融合模型与LASSO[9-10]变量筛选的能力。
2.软件实现及模拟实验评价指标
所有程序使用R语言编写实现,主要的R包有randomForest,RankAggreg[5],plsdepot,penalized pROC和mvtnorm等。融合模型与LASSO比较过程中,每种条件模拟100次,通过模型预测的灵敏度、特异度、预测准确性和AUC值等指标的平均值来评价两种方法。
3.模拟实验结果
模拟实验结果显示融合模型对差异变量的筛选要优于LASSO,实验结果见表1。当噪声固定为1000时,两种模型所筛选的差异变量数随所设差异变量和观测数的增加而增大;当所设差异变量和观测数较小时,融合模型所筛选的差异变量的AUC值高于LASSO;当所设差异变量和观测数较大时,融合模型和LASSO所筛选的差异变量的AUC值几乎相同;在相同的参数条件下,融合模型所筛选的差异变量数少于LASSO。
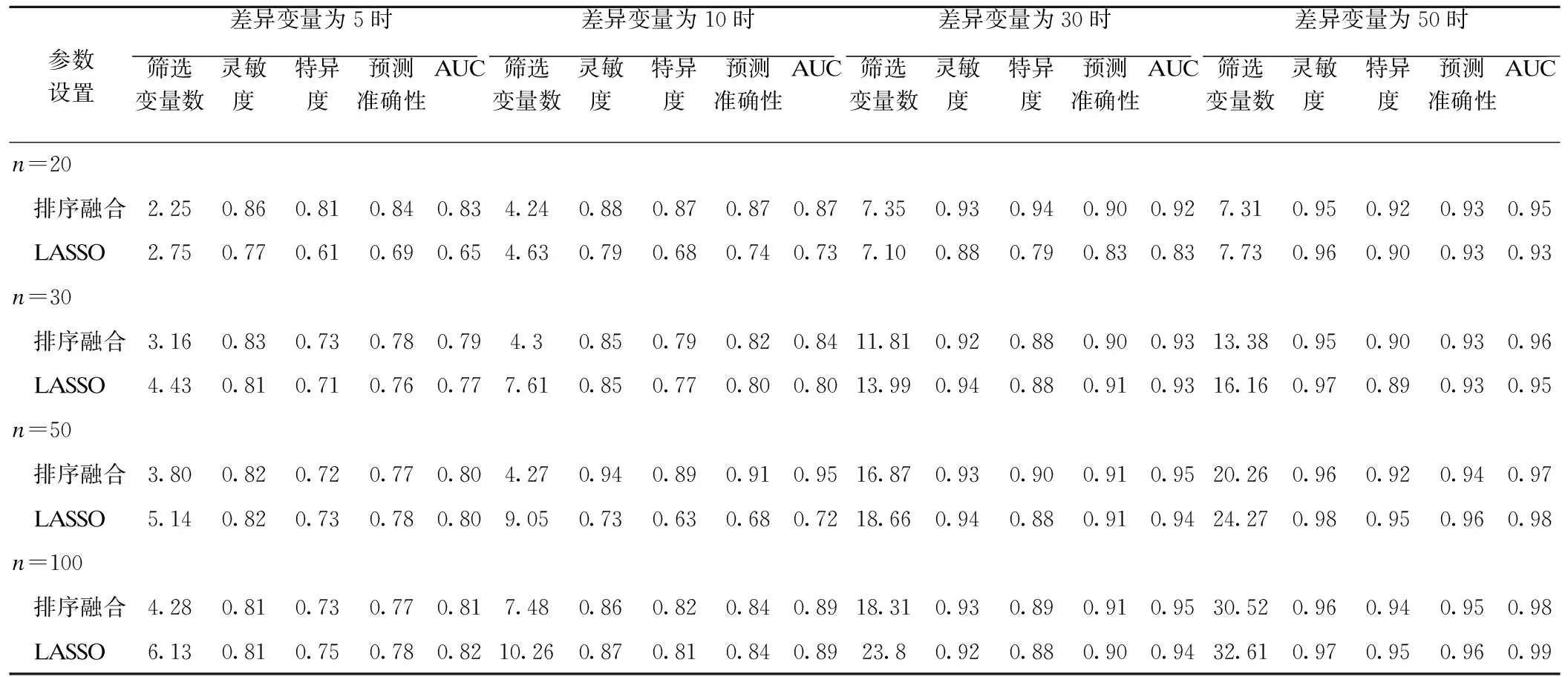
表1 差异变量分别为5,10,30,50时模拟实验结果
*:n为样本量,病例组和对照组相等;表中数据为100次模拟实验的均值。
实例应用
本研究选取儿童过敏性紫癜20例和紫癜性肾炎16例,进行非靶向代谢组学研究。测得过敏性紫癜和紫癜性肾炎血浆样品中小分子代谢物的代谢组学图谱,所包含特征变量数为536。
1.基于四种常见变量筛选方法生成融合序列
生成融合序列的四种常见变量筛选方法包括:t检验、Wilcoxon秩和检验、PLS和RForest,四种方法及排序融合模型变量重要性排序见表2。融合序列排名前五的物质标签为21,56,15,227和411。
2.利用融合序列筛选儿童紫癜型肾炎差异代谢物
融合模型差异变量筛选结果见图1,当排序融合模型中变量数达到12时,融合模型的预测准确率和AUC值最大,分别是0.89和0.96;灵敏度和特异度分别为1.00和0.80。
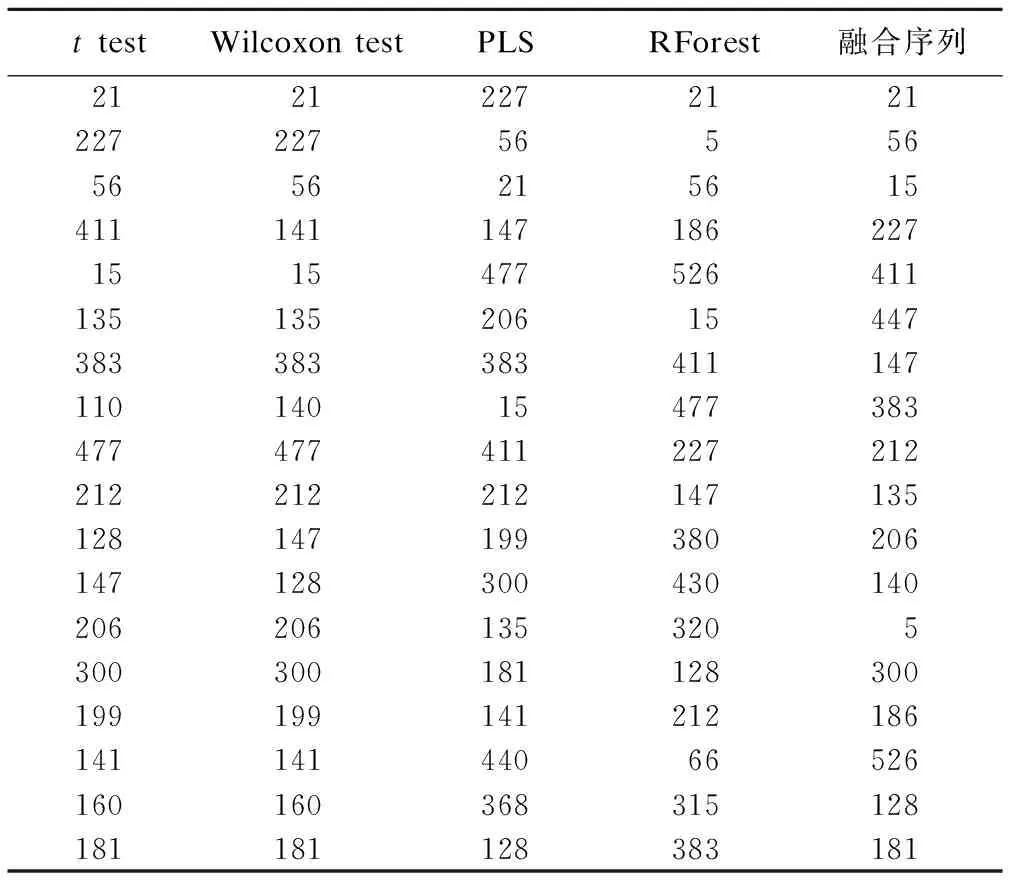
表2 四种常见变量筛选方法及排序融合的变量重要性排序
*:数值指某种小分子代谢产物。
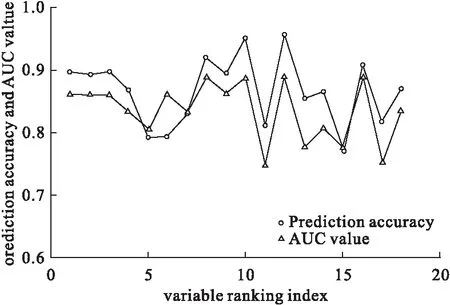
图1 Logistic模型预测的准确性和AUC值与引入模型变量数的关系*:变量按融合序列中重要性递减顺序进入模型
讨 论
对于高维组学数据,不同的分析方法对同一数据分析可能筛选出不同的差异变量,同时差异变量的排序也可能不同,选择某一种变量筛选方法结果具有一定的局限性。排序融合模型为高维组学数据分析提供了新的分析策略,能够结合多种现有组学分析方法,因此具有较强的适用性;由于各变量筛选方法的原理不同,某一变量筛选方法对特定数据具有较强的差异变量筛选能力,排序融合模型通过多种变量筛选方法的相互组合,充分发挥各单一变量筛选方法对代谢组学数据中不同特征子集差异变量的筛选优势,使分析结果能够取长补短,可靠性更好。
理论上只要变量筛选方法能够提供差异变量重要性排序,能够纳入排序融合模型的变量筛选方法在数量上不受限制,但考虑到模型的计算成本和同类变量方法过多导致排序融合模型变量筛选的偏性,建议同一类变量筛选方法只纳入一种。本篇论文结合了t检验、Wilcoxon秩和检验、PLS和Rforest,随着代谢组学的发展,基于不同原理的数据分析方法还将不断提出,这也为排序融合模型的拓展提供了方法学上的支持。
实际上代谢组学数据中大多数变量不满足正态分布,还可能存在离群值,尤其在观测数较小这种情况尤为突出。在模拟实验中将差异变量设为可能存在离群值的指数分布,融合模型能够筛选出较少的差异变量,获得较好的预测模型。特别是当差异变量数和观测数较小时融合模型所筛选出的差异变量的AUC值要大于LASSO对应的AUC值。排序融合模型对代谢组学数据差异变量的筛选具有较高的准确性,为代谢组学数据提供新的分析思路和方法。
