基于最长公共子序列的DNA序列相似性分析
2018-10-31吴东根周小安
吴东根, 周小安
(深圳大学 信息工程学院, 广东 深圳 518000)
引言
人类从未停止过对生物本质的好奇和探索,随着过去几十年来生物分子学以及基因技术的飞速发展,人类得到的生物信息呈现爆炸性的增长趋势。生物技术尤其是分子生物技术的快速发展,已经将人们带入一个新的时代——基因组时代。在人类基因组计划[1](Human Genome Project,简称HGP)的实施过程中,科学家们获得了海量的人体基因信息,这极大促进了生物学尤其是基因学的发展,让人类步入了后基因时代。
目前关于分析DNA相似性的方法已有很多,早期的有:Gibbs和Mclntre在1970年提出的点阵图分析方法[2]、傅里叶变换分析法[3]、Lipman和Pearson在1988年提出了FASTA算法[4]、Gatlin提出的条件熵方法[5];近期的有:三角函数法、谱动态法等等[6-11]。传统研究思路基本分为两个步骤:序列转换、转换后相似分析。在图形表示法中,第一步将DNA序列转化为图形模式,如转化为二维、三维坐标[12-13]中的曲线,第二步利用图形构造矩阵,再用矩阵的不变量进行DNA序列相似性分析;在数值表示法中,第一步将DNA序列转化为数值模式,如转化为二进制数或整数[14-15],第二步通过计算序列之间向量的欧氏距离或夹角余弦值[16]来分析DNA序列的相似性。
传统的方法存在一定的缺陷,都需要进行序列转换,这可能会在转换过程中丢失原始DNA序列的信息,并增加工作量。基于此,本文研究旨在寻找并研究一种不需要序列转换并且降低工作量的新方法,同时能达到相似性分析的要求。
1 基本原理
1.1 最长公共子序列
两个DNA序列之间的最长公共子序列越长,说明这两个DNA序列之间具有更多的相似性[1]。最长公共子序列(Longest Common Subsequence)是一个在一个序列集合中(通常为两个序列)用来查找所有序列中最长子序列的问题。这与查找最长公共子串的问题不同的地方是:子序列不需要在原序列中占用连续的位置 。最长公共子序列问题是一个经典的计算机科学问题,也是数据比较程序,比如Diff工具,和生物信息学应用的基础。最长公共子序列的定义如下:
一个序列S,如果分别是两个或多个已知序列的子序列,且是所有匹配此条件序列中最长的,则S称为已知序列的最长公共子序列[17]。
例如,序列1A2C3D4B56和B1D23CA45B6A的最长公共子序列为123456、12C4B6、12C456和1234B6(不一定只有一条),最长公共子序列长度为6。实验中只选取其中一条最长公共子序列即可。
求解序列的最长公共子序列问题首先需要求解动态规划表。设有两个序列:A=a[0]a[1]…a[m-1],A中任意一个元素记为a[i],i∈[0,m-1];B=b[0]b[1]…b[n-1],B中任意一个元素记为b[j],j∈[0,n-1]。A的长度为m,B的长度为n,生成大小为m×n的矩阵记为tr,tr的行数为m、列数为n。记tr[i][j]的含义为序列a[0]…a[i]与序列b[0]…b[j]的最长公共子序列的长度。从左到右,再从上到下计算矩阵tr中的各个元素。
(1)tr[i][0]的含义是a[0]…a[i]与b[0]的最长公共子序列长度。b[0]只有一个字符,所以tr[i][0]最大为1。如果a[i]=b[0],令tr[i][0]=1,一旦tr[i][0]被设置为1,之后的tr[i+1][0]、…、tr[m-1][0]也都为1。
(2)矩阵tr第一行与步骤1同理,如果a[0]=b[j],则令tr[0][j]=1,一旦tr[0][j]被设置为1,之后的tr[0][j+1]…tr[0][n-1]也都为1。
(3)对其它位置(i,j),tr[i][j]的值可能来自以下3种情况:
①可能是tr[i-1][j],代表a[0]…a[i-1]与b[0]…b[j]的最长公共子序列长度。
②可能是tr[i][j-1],代表a[0]…a[i]与b[0]…b[j-1]的最长公共子序列长度。
③如果a[i]=b[j],还可能是tr[i-1][j-1]+1。
在这3个可能值中,选取最大的作为tr[i][j]的值即可。
tr矩阵中右下角的元素值代表A和B的最长公共子序列的长度。通过整个tr矩阵的状态,可以得到最长公共子序列。具体方法如下:
步骤1从矩阵的右下角开始,有3种移动方式:向上、向左、向左上。假设移动的过程中,用p表示此时的行数,q表示此时的列数,用一个变量C(其中C=c[0]c[1]…)来表示最长公共子序列。
步骤2如果tr[p][q]大于tr[p-1][q]和tr[p][q-1],说明之前计算tr[p][q]的时候,一定是选择了决策tr[p-1][q-1]+1,可以确定a[p]等于b[q],并且这个字符一定属于最长公共子序列,把这个字符放进C,然后向左上方移动。
步骤3如果tr[p][q]等于tr[p-1][q],说明之前在计算tr[p][q]的时候,tr[p-1][q-1]+1这个决策不是必须选择的决策,向上方移动即可。
步骤4如果tr[p][q]等于tr[p][q-1],与步骤3同理,向左方移动。
步骤5如果tr[p][q]同时等于tr[p-1][q]和tr[p][q-1],向上移动或向左移动都可以。
通过上面的方法,就可以用C++程序得到两个序列其中一条最长公共子序列,并得到这条最长公共子序列的长度。
1.2 相似系数
本文将一个DNA序列视为以{A、G、T、C}为符号集的信号序列,记x、y为物种的名称,如mouse、human等等,其中物种名称x与物种名称y可以相同。记d(x)为x物种的DNA序列的长度,可以定义两物种DNA序列中长度更大的那条序列的长度D(x,y):
D(x,y) = max{d(x),d(y)}
(1)
记B(x,y)为两个物种DNA序列的最长公共子序列长度,其中B(x,y)≤D(x,y)。可以定义两个物种的相似系数H(x,y),用来评判两物种的相似程度:
(2)
其中0≤H(x,y)≤1,H(x,y)越接近1,表示x、y两物种相似度越高,当H(x,y)等于1时,表示两个物种是同一物种;H(x,y)越接近0,表示x、y两物种相似度越低,当H(x,y)等于0时,表示两个物种是不同物种。
2 分析对象
本文从NCBI数据库(http://www.nubi.nlm.nih.gov)中找到并下载了7种病毒的DNA数据,表1中列出了本文中所需要研究的7种DNA序列片段的信息。表2引用文献[1]的10个物种的β-globin基因第一外显子序列信息,其中序列的长度为86到105不等。

表2 10种β-globin基因第一外显子序列
3 实验结果
3.1 两物种DNA序列的最长公共子序列
根据前文所描述求解最长公共子序列的方法,可以求出两个物种的最长公共子序列,称为物种对的最长公共子序列。表3只列出了一部分物种对的最长公共子序列,每个物种对的最长公共子序列可能不止一条,仅选取其中任意一条即可。其它物种对的最长公共子序列的求法一样,在此不一一列出。
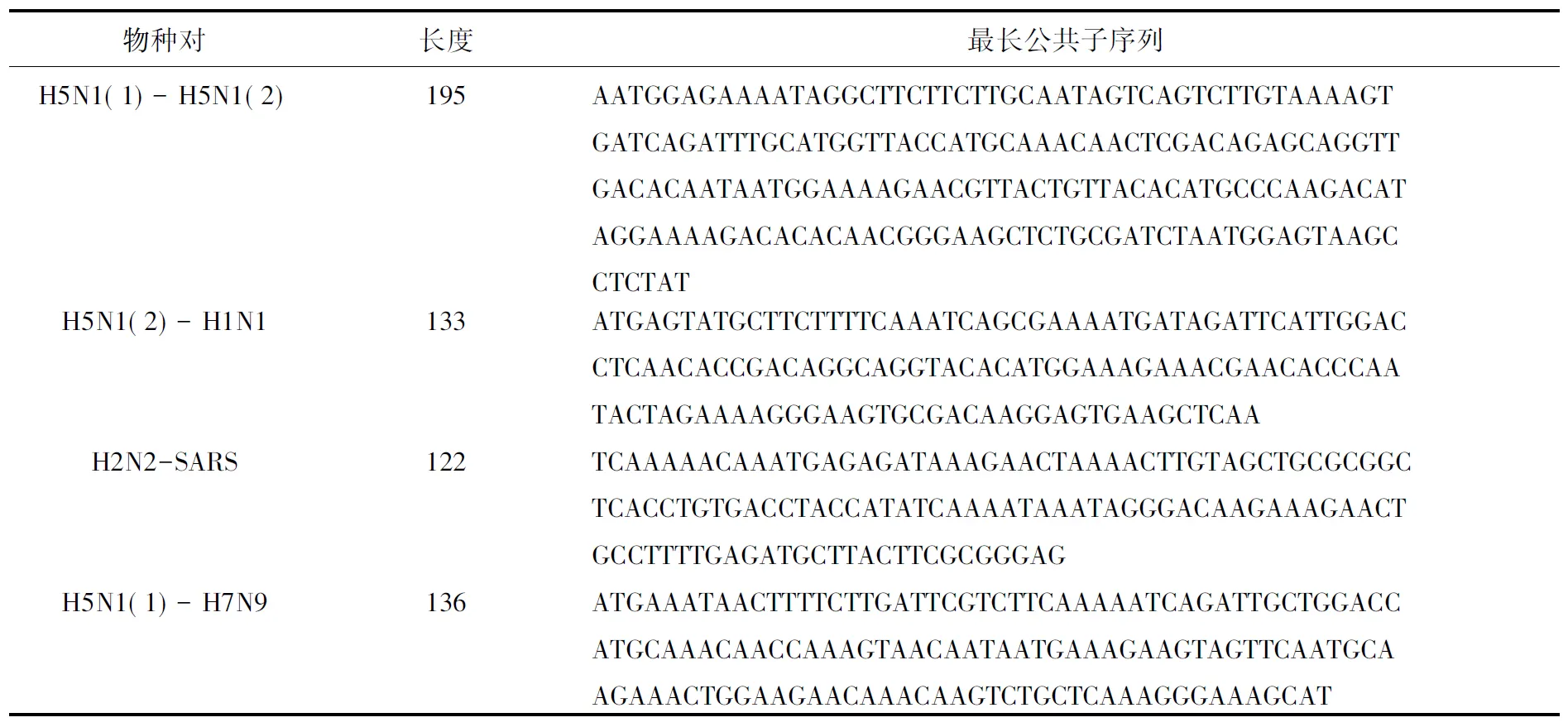
表3 两物种DNA序列的最长公共子序列
3.2 7种病毒DNA序列实验情况
利用最长公共子序列算法,求出7种病毒DNA序列两两间的最长公共子序列,利用定义的相似系数,求出7种病毒DNA序列的相似系数,所得相似系数见表4。
观察表4中的数据, H5N1(1)与H5N1(2)的相似系数是0.975,非常接近1,说明H5N1(1)与H5N1(2)相似性很高。这与实际结论一致,相似系数分析结果与事实符合,说明基于最长公共子序列的方法对判断DNA序列的相似性有效。
3.3 10种β-globin基因第一外显子序列实验情况
对文献[1]中10种不同β-globin基因第一外显子序列进行了实验,计算出各物种对的相似系数,所得数据见表5。
观察表5中的数据,mouse-rat物种对、human-gorilla对和goat-bovine对的相似系数比较高,都十分接近1。在实际中,mouse和rat为鼠类,其DNA序列应该具备最大的相似性,实验结果与实际情况相符;human和gorilla的相似系数最为接近1,即相似性最大,与实际情况相符。其次发现物种gallus和物种opossum分别与其他9个物种的相似系数比较小,这也是与实际相符的。证明本方法对相似性分析是有效的。另一方面,human-mouse对、mouse-gorilla对和gorilla-bovine对的相似系数接近0.9,比起其它物种对的相似系数,这些物种对更靠近1,说明这些物种对有很大的相似性,这与实际情况不符。从生物学的角度来看gorilla-bovine、gorilla属于灵长目,bovine属于偶蹄目,不是同源物种,不应该有很大的相似性,因此实验显示的数据是有偏差的。也许是由于这些物种在某些功能或某些结构中存在相似性。在其它的文献里也曾出现过类似的情况,见文献[18]。

表4 7种病毒DNA序列相似系数矩阵
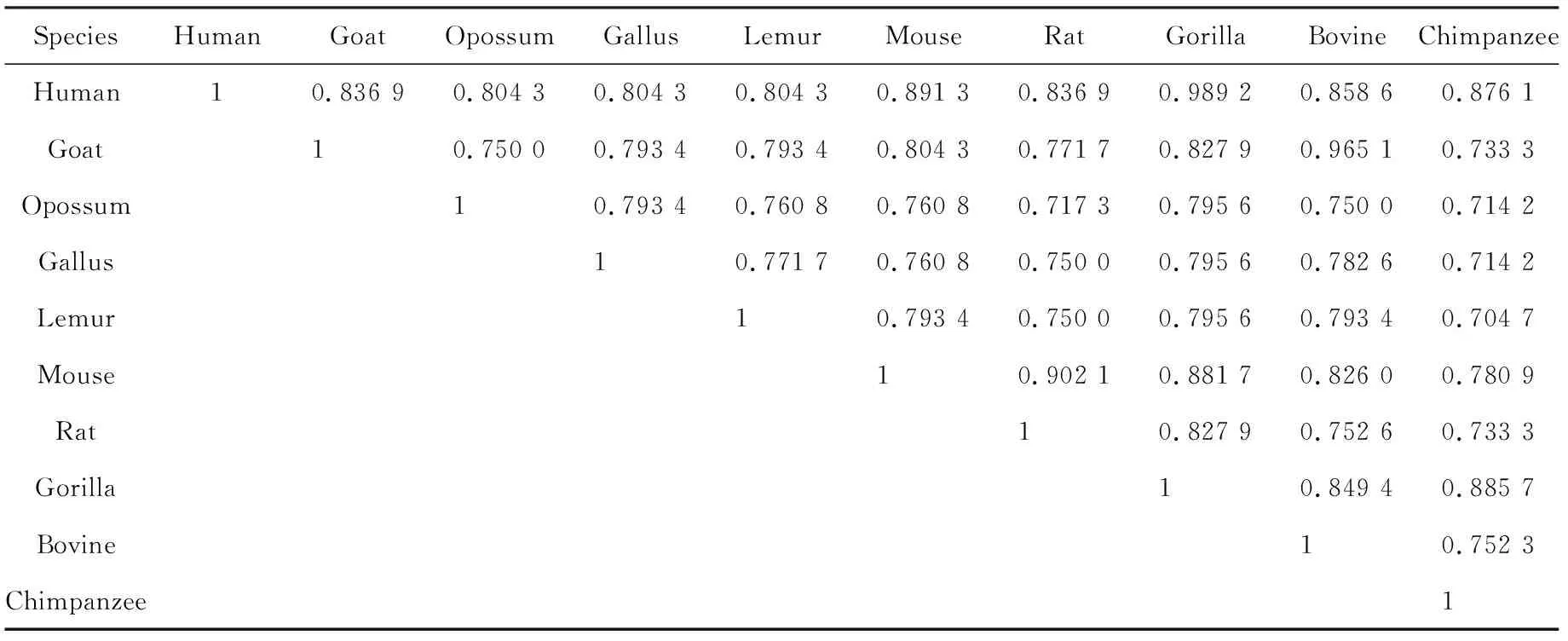
表5 10种β-globin基因第一外显子序列相似系数矩阵
将表4与表5数据对比,可以发现,当处理的DNA序列长度较大时,相似系数会更加准确,不同物种的相似系数会更加趋向0,同源物种的相似系数会更趋向1,这是本方法的优势;相比之下,处理的DNA序列较短时,优势就没那么明显,会出现不同物种的相似系数也十分趋近1的情况。另外,本文的方法既可用作相同长度DNA序列的相似性分析;也可以用作长度不同的DNA序列的相似性分析,这也是该方法的优势。
4 结束语
本文提出一种基于最长公共子序列的DNA序列相似性分析方法,并通过实验证明了此方法的有效性,同大多数传统的方法相比,本方法无需进行DNA序列的图形转换或数值转换,直接从原始DNA序列出发,大大降低了工作量。且方法既可以对比长度相等的DNA序列相似性,也可以对比长度不相等的DNA序列相似性。处理的序列长度越大,评判相似性的程度越好。同时也为DNA序列相似性分析增添了一种新的研究方法。本文只使用了其中一条最长公共子序列,是否能通过求得所有最长公共子序列进而加深相似性值得进一步研究。
