1例先天性肾上腺发育不良的诊断
2018-10-26王会贞李文静杨海花陈永兴卫海燕
王会贞,李文静,杨海花,陈永兴,卫海燕
(郑州大学附属儿童医院/河南省儿童医院/郑州儿童医院,郑州450052)
先天性肾上腺发育不良(adrenal hypoplasia congenita,AHC) 是一种罕见的与肾上腺皮质发育有关的遗传性疾病,1948年由Sikl首先报道。AHC由编码核受体蛋白的NR0B1基因突变或完全缺失引起,以X染色体隐性方式遗传,女性携带致病基因,男性发病。NR0B1基因也称剂量敏感-性反转-肾上腺发育不良基因1,定位于染色体Xp21,包括2个外显子和1个内含子。NR0B1基因存在于垂体分泌促性腺激素的细胞和下丘脑核团中[1],在合成类固醇激素的内分泌腺体中表达[2],主要调节肾上腺和性腺发育、分化、激素的合成与分泌。因此,NR0B1基因突变可导致原发性糖皮质激素及盐皮质激素缺乏,婴儿期及儿童早期多表现为失盐症状,易误诊为先天性肾上腺皮质增生症(congenitial adrenal hyperplasia,CAH)、醛固酮缺乏症及原发性慢性肾上腺皮质功能减退症(Addison病)等。现对1例确诊AHC患儿的临床资料及基因检测结果进行分析,以探讨AHC的有效诊断方法。
1 资料分析
患儿男,6岁8个月,因“生长迟缓6 a”于2015年9月29日入院。患儿出生8个月大时曾因皮肤色素沉着、高钾血症、低钠血症、休克就诊,诊断为CAH,口服氢化可的松、氟氢可的松治疗至今,病情好转,期间未复诊。否认家族中有遗传代谢疾病病史,无其他异常表现。本次入院查体:身高111.1 cm,体质量23 kg,体温36.5 ℃,心率102次/min,呼吸25次/min,血压84/58 mmHg;患儿神志清,精神一般;皮肤光滑,无明显色素沉着;心肺腹未见异常;双侧睾丸体积约1 mL,阴茎长约4 cm,阴毛发育为PH1期;神经系统查体未见异常。实验室检查:血清17-羟孕酮(17-OHP)0.261 ng/mL、促肾上腺皮质激素释放激素(ACTH)161.7 pg/mL、皮质醇8.708 ng/mL、睾酮(T)<0.025 ng/mL、孕酮(P)<0.030 ng/mL。生长激素(GH)激发试验:GH峰值13.90 ng/mL、胰岛素样生长因子-1(IGF-1)44.235 ng/mL。肾素-血管紧张素-醛固酮检测:血清血管紧张素Ⅰ 4.498 ng/mL、肾素活性0.48 ng/(mL·h)、血管紧张素Ⅱ92.018 pg/mL、醛固酮49.914 pg/mL。血清电解质检测:钾3.90 mmol/L、钠138.0 mmol/L、氯103.0 mmol/L。心肌酶检测及血气分析均无异常。甲状腺功能无异常。MRI显示垂体高约4.2 mm。腹部彩超检查结果示肝脏、胰腺、脾脏、肾脏未见异常,双侧肾上腺区未见明显占位。X线检测结果显示患儿骨龄相当于7岁。结合病史及本次检查结果,拟诊为AHC。征得患儿家长同意,对患儿进行NR0B1基因检测:采患儿静脉血5 mL,用Qiagen公司试剂盒提取基因组DNA,将DNA进行片段化,然后进行DNA建库,对目标DNA进行捕获,用Illumina Hiseq X Ten高通量测序仪进行测序,测序长度为2×150 bp。对测序仪获取的原始数据进行比对,对变异位点注释,筛选出疑似片段缺失性有害突变,进行PCR扩增,患儿NR0B1基因的1号外显子(E1)和2号外显子(E2)均未扩增出条带(见图1)。进一步用多重连接探针扩增技术(MLPA)验证,结果显示患儿NR0B1 基因E1、E2区域存在大片段缺失(见图2),结合临床资料,AHC诊断明确。患儿母亲NR0B1 基因的MLPA检测结果显示,未检测到NR0B1基因缺失/重复(见图3),提示该患儿为新生NR0B1基因突变可能。
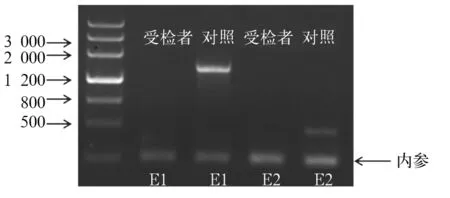
注:患儿NR0B1基因的E1和E2均未扩增出条带。
图1患儿NR0B1基因的PCR扩增结果
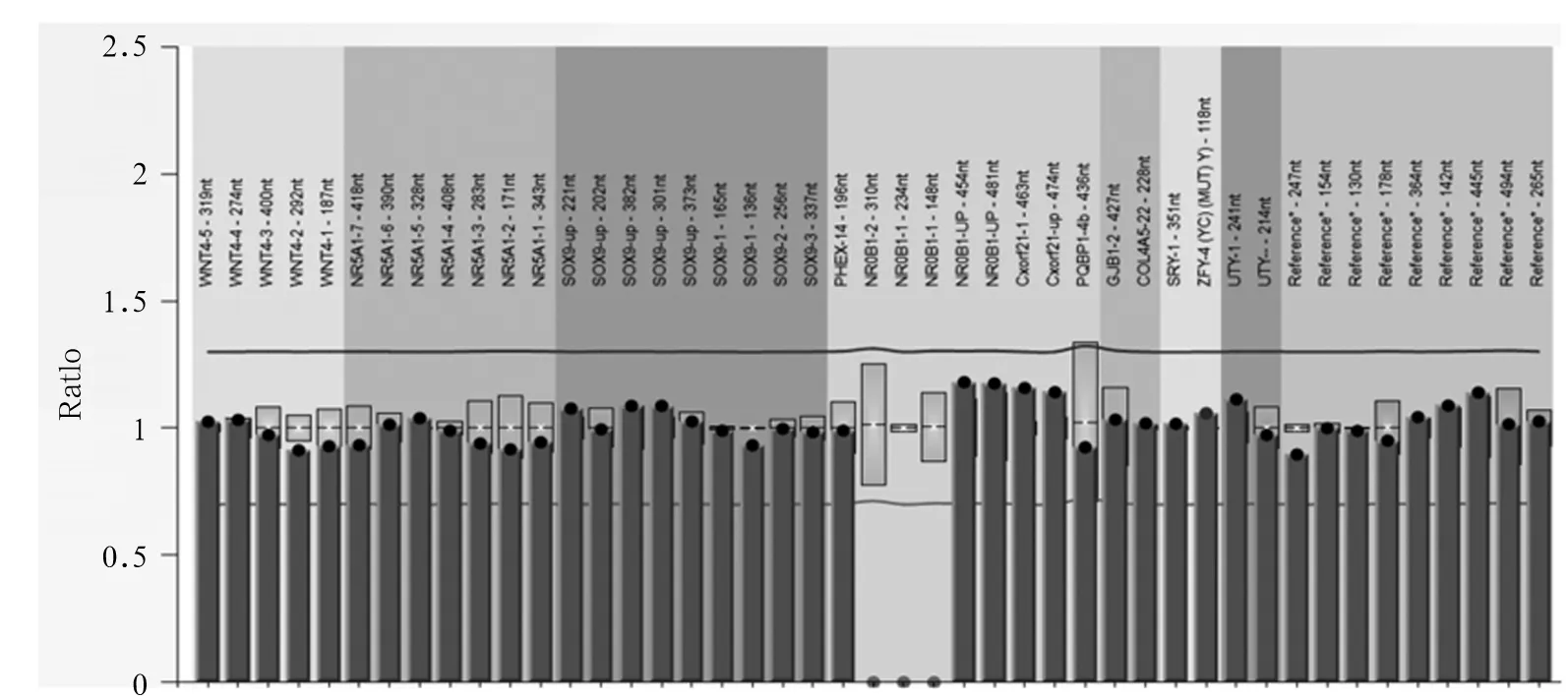
注:患儿NR0B1基因E1和E2区域存在大片段缺失。
图2患儿NR0B1基因的MLPA检测结果

注:患儿母亲NR0B1基因未检测到缺失/重复。
图3患儿母亲NR0B1基因的MLPA检测结果
2 讨论
AHC是一种罕见的由编码核受体蛋白的NR0Bl基因突变或完全缺失引起,以X染色体隐性方式遗传。NR0Bl基因定位于染色体Xp2l,包括2个外显子和1个内含子,编码470个氨基酸,属于孤核受体超家族成员。近年来,随着分子诊断技术的进步和对AHC研究的深入,基因检测为该疾病的诊断提供了新的手段。到目前为止,大约有200余种NR0B1基因突变在国内外被发现,突变的类型包括碱基缺失、碱基插入、剪切突变、无义突变、移码突变、错义突变、大片段的缺失和重组等[3,4],报道最多的是移码突变和无义突变,少数为错义突变、单基因缺失或碱基插入。NR0Bl基因突变大多位于基因编码区,或影响蛋白折叠,或造成蛋白结构改变,最终导致蛋白被截断,影响功能。
AHC临床表现复杂,婴幼儿期发病者多伴有体质量不增或下降、拒食、呕吐、腹泻、脱水、嗜睡、皮肤色素沉着等表现,血液生化检查结果常提示严重的高钾、低钠、酸中毒,与CAH、醛固酮减少症、Addison病表现有所重叠,容易误诊[5~9]。丁宇等[10]报道3例新生儿期起病、1例婴儿期起病患儿,均表现为难以纠正的低钠、高钾血症,依据患儿的临床资料及基因检测结果,最终分别确诊为CAH、AHC、假性醛固酮减少症I型、醛固酮减少症。有研究[11]报道1例出生21 d的男婴,因喂养困难、体质量增长不良就诊,查体皮肤色素沉着、外生殖器正常;实验室检查结果显示高钾、低钠、低血糖,皮质醇水平降低、ACTH水平升高,17-OHP水平轻度增高,11-脱氧皮质醇水平显著升高,临床诊断为CAH。基因检测结果提示NROB1基因E2错义突变,第447位氨基酸由脯氨酸变为亮氨酸,最终确诊为AHC,NROB1基因检测结果显示患儿母亲为携带者。另有研究[12]报道1例8岁男孩因腹痛、呕吐、腹泻、皮肤色素沉着就诊,实验室检查结果显示高血钾、低血钠,血清皮质醇水平降低、ACTH水平升高、17-OHP水平正常,诊断为Addison病,直到12岁因性腺发育不良就诊时考虑AHC;基因检测结果提示患儿NR0B1基因E1半合子突变,为移码突变,使多肽链提前终止,导致蛋白功能受损,最终确诊为AHC。Bizzarri等[8]分析10年间就诊的51例低钠血症患儿的疾病谱,结果显示9例是因长期钠离子消化道、肾脏丢失或摄入不足引起,10例为中枢神经系统疾病,19例为CAH,4例为AHC,2例为醛固酮合成酶缺陷,1例为假性醛固酮减少症,2例为家族性糖皮质激素缺乏,2例为肾脏畸形,2例病因未明。由此可见,仅根据临床表现、生化检查结果明确病因诊断是非常容易误诊的。在临床工作中对存在皮肤色素沉着、高钾、低钠、ACTH水平明显升高的患儿应谨慎诊断,可进行NR0B1基因检测。
本例患儿最初发病时表现为皮肤色素沉着、电解质紊乱(高钾血症、低钠血症)、休克,诊断为CAH,口服氢化可的松、氟氢可的松治疗,病情好转,但之后出现生长落后。本次入院检查发现,患儿血清ACTH水平升高,17-OHP、P、T水平均无明显升高,骨龄正常,CT检查肾上腺未见异常,与CAH临床表现及实验室检查结果不符。回顾分析患儿首次就诊时存在肾上腺皮质功能不全表现,之后出现生长落后,无骨龄超前等性早熟表现,考虑AHC可能。MLPA基因检测结果显示,患儿NR0B1基因E1、E2区域存在大片段缺失,为人类基因组数据库报道的已知致病性突变,AHC诊断明确。对其母亲进行NR0B1基因检测,未发现患儿母亲存在NR0B1基因缺失/重复,提示该患儿NR0B1基因的突变为新生突变。对可疑AHC患儿及其母亲进行NR0B1基因检测,将有助于AHC的确诊,也有助于基因突变来源、是否为新生突变的判断;如果常规PCR基因检测无异常发现,应进一步实施MLPA基因检测。
综上所述,AHC临床表现复杂、多变,婴幼儿发病者多伴有体质量不增或下降、拒食、呕吐、腹泻、脱水、皮肤色素沉着、电解质紊乱等表现。NR0B1基因检测为AHC的确诊提供了可靠手段,对患者早期诊断和规范治疗有重要意义。
