HPLC法测定麝香接骨丸中羟基红花黄色素A的含量
2018-10-08戴作波姜志戎
戴作波,姜志戎
江苏省宝应县中医医院药剂科,江苏宝应, 225800
麝香接骨丸是该院自行研制的纯中药制剂,由红花、血竭、麝香、丁香、骨碎补、自然铜、当归、杜仲等十五味中药组成。有和营活血接骨续筋功效。用于跌打损伤所致软组织损伤、骨折脱位中后期筋肉挛痛见上述症候者。
1 仪器与试剂
1.1 仪器
Waters Alliance e2695型高效液相色谱仪,Waters 2998型PDA检测器(美国Waters公司)。METTLER TOLEDO MS-105DU型电子分析天平(0.01 mg,瑞士梅特勒-托利多公司)。KH-500DB型数控超声波清洗器 (昆山禾创超声仪器有限公司)。Milli-Q DIRECT8型纯水仪(美国Millipore公司)。
1.2 试药
麝香接骨丸(批号:170612,170626,170701,由本院提供),羟基红花黄色素A对照品(批号:111637-201609,纯度91.9%,购自于中国食品药品研究检定院)。甲醇(色谱纯,美国TEDIA公司),磷酸(色谱纯,上海阿拉丁生化科技股份有限公司)。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent Extend C18色谱柱(250 mm×4.6 mm,5 μm);流动相:甲醇-0.7%磷酸溶液(30:70);流速:1.0 mL/min;检测波长:403 nm;柱温:30℃;进样量:10 μL。
2.2 提取条件的考察
该方中红花为饮片粉末入药,因此在考察提取条件时参考2015年版《中国药典》“红花”项下的有关内容:取本品粉末(过三号筛)约0.4 g,精密称定,置具塞锥形瓶中,精密加入25%甲醇50 mL,称定重量,超声处理(功率 300 W,频率 50 kHz)40 min[1],放冷,再称定重量,用25%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
据此考察了甲醇、75%甲醇、50%甲醇、25%甲醇和水为提取溶剂,30 min、40 min、50 min 和 60 min 为提取时间[2],对制剂中羟基红花黄色素A提取得率的影响。

表1 提取条件考察
由此可知,以25%甲醇为提取溶剂,超声处理40 min,效果较好,见表 1。
2.3 线性关系考察
精密称取羟基红花黄色素A对照品2.782 mg,置10 mL量瓶中,加流动相溶解并稀释至刻度,摇匀,即得对照品贮备液。精密量取对照品贮备液4 mL,置10 mL量瓶中,加流动相稀释至刻度,摇匀,即得6#对照品溶液;精密量取对照品贮备液2 mL,置10 mL量瓶中,加流动相稀释至刻度,摇匀,即得5#对照品溶液;精密量取对照品贮备液1 mL,置10 mL量瓶中[3],加流动相稀释至刻度,摇匀,即得4#对照品溶液;精密量取对照品贮备液0.5 mL,置10 mL量瓶中,加流动相稀释至刻度,摇匀,即得3#对照品溶液;精密量取3#对照品溶液5 mL,置10 mL量瓶中,加流动相稀释至刻度,摇匀[4],即得2#对照品溶液;精密量取2#对照品溶液5 mL,置10 mL量瓶中,加流动相稀释至刻度,摇匀,即得1#对照品溶液。精密吸取上述溶液各10 μL,注入液相色谱仪,测定,记录峰面积。每份溶液平行进样2次,求平均峰面积。以平均峰面积为纵坐标(Y),对照品溶液浓度(μg/mL)为横坐标(X),进行线性回归[5]。

表2 标准曲线
羟基红花黄色素A的回归方程为Y=259 81 X-205 03(r=0.999 7),线性范围为 3.20~102.27 μg/mL,见表2。
2.4 定量限与检测限试验
以S/N≈10为标准,羟基红花黄色素A的定量限为 1.60 μg/mL;以 S/N≈2~3 为标准,羟基红花黄色素A的检测限为0.40 μg/mL。
2.5 重复性试验
取批号为170626的本品6份,研细,每份取约0.5 g,精密称定,置具塞锥形瓶中,精密加入25%甲醇25 mL,密塞,称定重量,超声处理(功率250 W,频率50 kHz)40 min,放冷,再称定重量[6],用 25%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。精密吸取上述溶液各10 μL,注入液相色谱仪,测定,记录峰面积。每份溶液平行进样2次,求平均峰面积。以平均峰面积照标准对照法计算含量[7]。

见表3表4。

表3 标准对照

表4 重复性试验
2.6 精密度试验
取“重复性试验”中第1份供试品溶液为研究对象。精密吸取该溶液10μL,注入液相色谱仪,测定,记录峰面积。连续进样6次,计算平均峰面积和RSD,见表5。
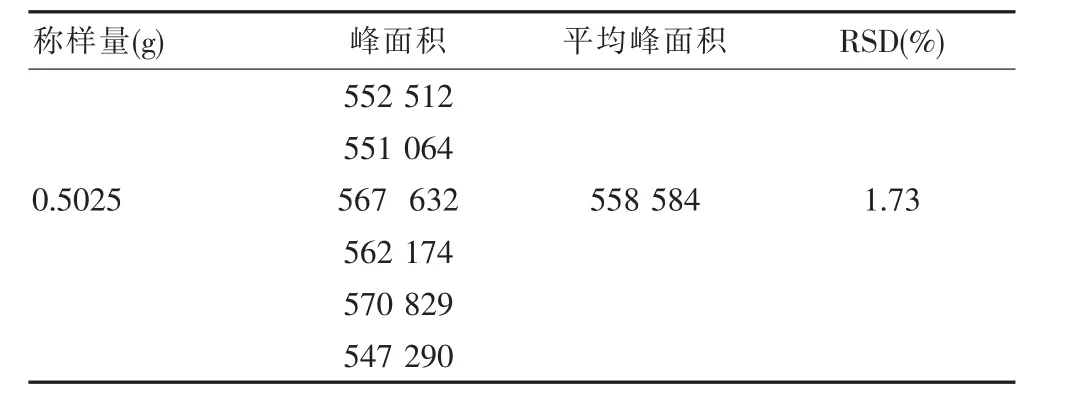
表5 精密度试验
2.7 稳定性试验
取“重复性试验”中第1份供试品溶液为研究对象。精密吸取该溶液10 μL,注入液相色谱仪,测定,记录峰面积。间隔若干时间进样1次,计算平均峰面积和RSD,见表6。
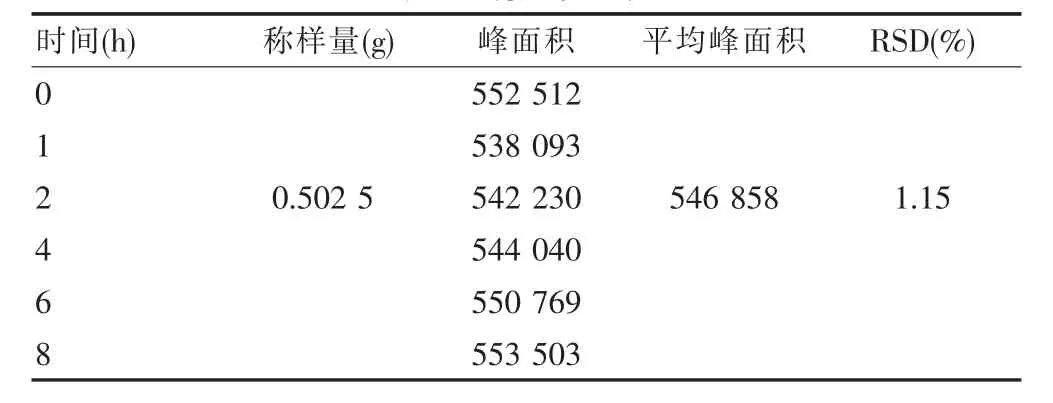
表6 稳定性试验
2.8 回收率试验
取批号为170 626的该品6份,研细,每份取约0.25 g,精密称定,置具塞锥形瓶中,精密加入羟基红花黄色素 A 对照品溶液(265.59 μg/mL)1 μL 和 25%甲醇 24 μL,密塞,称定重量[8],超声处理(功率 250 W,频率50 kHz)40 min,放冷,再称定重量,用25%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。精密吸取上述溶液各10 μL,注入液相色谱仪,测定,记录峰面积。每份溶液平行进样2次,求平均峰面积。以平均峰面积照标准对照法计算[9-10],见表7、表8。


表7 标准对照

表8 回收率试验
2.9 含量测定
取批号为 170 626、170 612、170 701 的本品,每批3份,研细,每份取约0.5 g,精密称定,置具塞锥形瓶中,精密加入25%甲醇25 mL,密塞,称定重量,超声处理(功率 250 W,频率 50 kHz)40 min,放冷,再称定重量,用25%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。精密吸取上述溶液各10 μL,注入液相色谱仪,测定,记录峰面积。每份溶液平行进样2次,求平均峰面积。以平均峰面积照标准对照法计算含量。批号为170 626的样品采用“重复性试验”前3份数据见表9、表10。
表9 标准对照
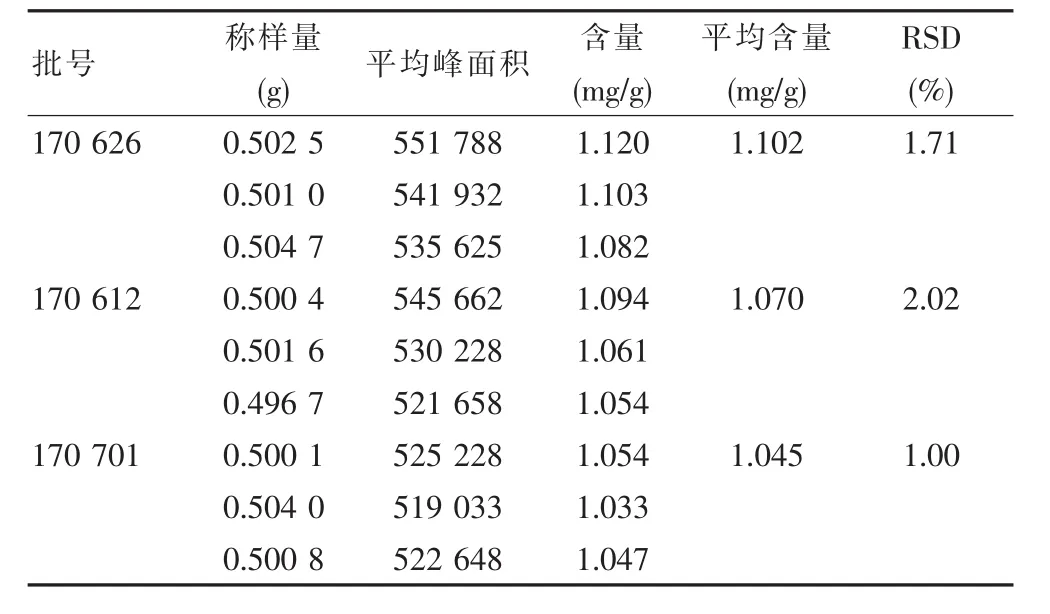
表10 含量测定
2.10 中间精密度
由该实验室另一名操作人员B,使用Kromasil C18 色谱柱 (250 mm×4.6 mm,5 μm), 以批号为170626的样品为研究对象,取3份,研细,每份取约0.5 g,精密称定,置具塞锥形瓶中,精密加入25%甲醇25 mL,密塞,称定重量,超声处理(功率250 W,频率 50 kHz)40 min,放冷,再称定重量,用 25%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。精密吸取上述溶液各10 μL,注入液相色谱仪,测定,记录峰面积。每份溶液平行进样3次,求平均峰面积。以平均峰面积照标准对照法计算含量,并与“重复性试验”前3份数据比较,见表11、表12。

表11 标准对照
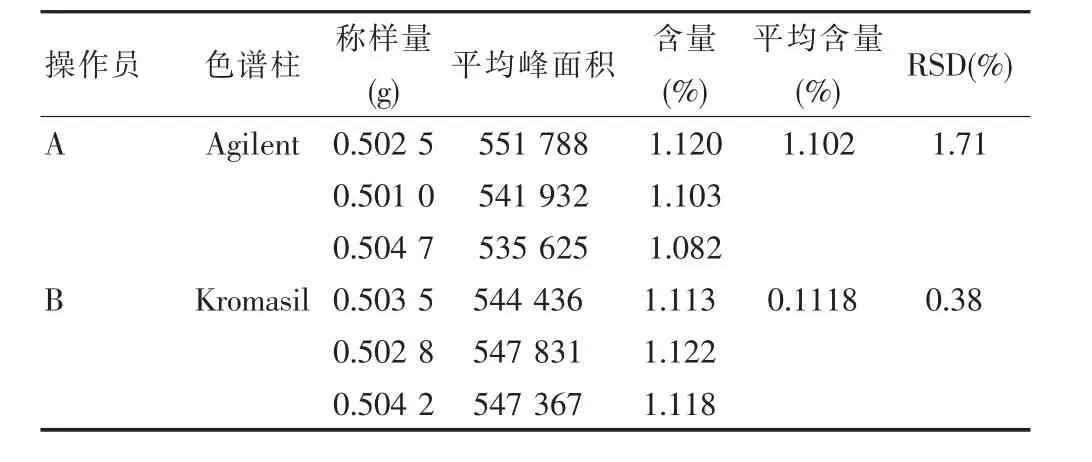
表12 中间精密度
2.11 含量限度
批号为170 612、170 626、170 701的该品三批制剂中,羟基红花黄色素A的平均含量分别为1.102 mg/g、1.070 mg/g、1.045 mg/g,三批的平均含量为 1.072 mg/g。考虑到原料药材产地、种植习惯、采收加工方法的差异和炮制方法的差异等因素拟以“平均含量×80%”为含量限度,即不低于0.85 mg/g。见图1。
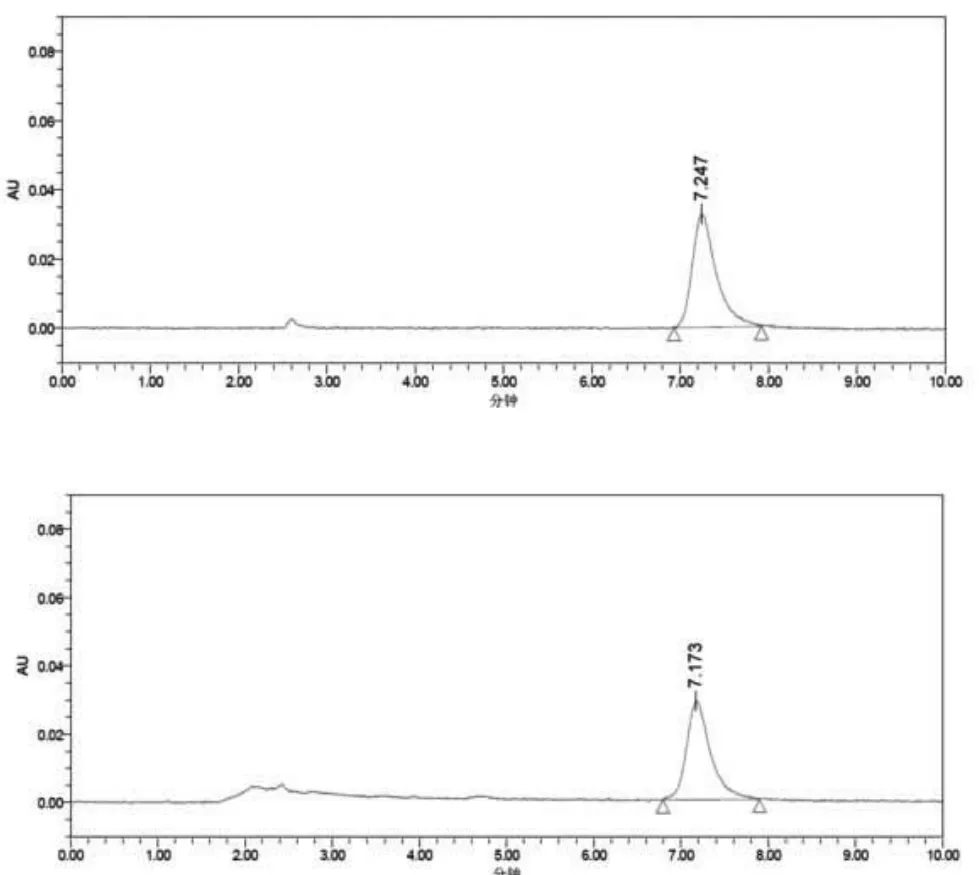
图1 含量限度
3 讨论
3.1 耐用性试验
该试验的色谱条件参考2015年版 《中国药典》“红花”项下相关内容。为配制方便,将流动相从“甲醇-乙腈-0.7%磷酸溶液 (26:2:72)” 改为 “甲醇-0.7%磷酸溶液(30:70)”。在此基础上,采用供试品溶液进行了耐用性试验,考察了色谱柱温度 (28℃、32℃)、流速(1.05 mL/min、0.95 mL/min)、波长(401 nm、405 nm)、流动相组成比例(32:68、28:72)、进样量(9 μl、11 μl)对色谱分离的影响。结果发现,上述条件的微小变动,对该试验中羟基红花黄色素A的测定未造成影响。
3.2 空白试验
为考察该方中其它中药饮片对红花中羟基红花黄色素A的测定是否存在干扰,该试验将红花从处方中除去,其余饮片仍按照【处方】组成比例和制剂工艺制成缺红花的空白制剂,并按照【含量测定】方法制成供试品溶液,依法测定分析。结果发现,该方中除红花外的其它饮片,对制剂中羟基红花黄色素A的测定无干扰。
4 讨论
采用高效液相色谱法测定主药红花中有效成份羟基红花黄色素A的含量,准确、简便,阴性无干扰,可作为本制剂的质量标准控制。HPLC法对红花中的活性成份羟基红花黄色素A进行测定。结果表明,该方法简便、准确、重现性好,可作为本制剂的质量标准控制。
