Electronic Stability of Eight-electron Tetrahedral Pd4 Clusters
2018-09-10SHENYanfangCHENGLongjiu
SHEN Yanfang , CHENG Longjiu ,2,*
1 School of Chemistry and Chemical Engineering, Anhui University, Hefei 230601, P. R. China.
2 Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Hefei 230601, P. R. China.
Abstract: Motivated by the unusual structure of the[Pd4(μ3-SbMe3)4(SbMe3)4] cluster, which is composed of a tetrahedral (Td) Pd(0) core with four terminal SbMe3 ligands and four triply bridging SbMe3 ligands capping the four triangular Pd3 faces (J. Am. Chem.Soc. 2016, 138, 6964), we performed a computational study of the structure and bonding characteristics of the Td [Pd4(μ3-SbH3)4(SbH3)4] cluster and a series of its analogues. The Td structure of the [Pd4(μ3-SbH3)4(SbH3)4] cluster could be explained by the cluster electron-counting rules based on the 18-electron rule for transition-metal centers; each sp3 hybridized Pd atom contributed ten valence electrons, and eight valence electrons were provided by one terminal SbH3 and three bridging μ3-SbH3 ligands. The[Pd4(μ3-SbH3)4(SbH3)4] cluster had a count of 104 valence electrons in total; chemical bonding analysis indicated that the cluster featured twenty electron lone pairs generated by d orbital of the four Pd atoms, twenty-four Sb―H σ bonds, four terminal Pd―Sb σ bonds, and four delocalized bonds. There were two bonding patterns of the eight delocalized electrons between the four capping Sb atoms and the Pd4 core. The first pattern was based on the superatom-network (SAN) model,whereby the palladium cluster could be described as a network of four 2e– superatoms. The second pattern was based on the spherical jellium model, whereby the cluster could be rationalized as an 8e– [Pd4(μ3-SbH3)4] superatom with 1S21P6 electronic configuration. The density functional theory (DFT) calculations showed that the Td [Pd4(μ3-SbH3)4(SbH3)4]cluster had a large HOMO-LUMO (HOMO: highest occupied molecular orbital; LUMO: lowest unoccupied molecular orbital) energy gap (2.84 eV) and a negative nucleus-independent chemical shift (NICS) value (-12) at the center of the[Pd4(μ3-SbH3)4(SbH3)4] cluster, indicating its high chemical stability and aromaticity. Furthermore, the NICS values in the range of 0–0.30 nm of the [Pd4(μ3-SbH3)4] motifs were much more negative than those of [Pd4(SbH3)4] in the same range,revealing that the overall stability of [Pd4(μ3-SbH3)4(SbH3)4] was likely derived from the local stability of Pd4(μ3-SbH3)4.Meanwhile, the d10···d10 interaction played a critical role in stabilizing the Pd4 tetrahedron structure, which is similar to the aurophilicity in Au-Au clusters. It was also found that there is a large difference in the stability of transition metal and non-transition metal clusters with a tetrahedron structure. The structures and bonding patterns of the designed analogues were similar to those of [Pd4(μ3-SbH3)4(SbH3)4]. To summarize, this study was relevant for deciphering the nature of the bonds in a tetrahedral complex with four cores and eight ligands, and predicting a series of analogues. It is expected that this work will provide more options for the synthesis of tetrahedral 4-core transition metal compounds.
Key Words: Metal cluster; Super valence bond; Superatom; Chemical bonding analysis; Closed-shell interaction;Aromaticity
1 Introduction
Clusters composed of atoms display unique physical,chemical and electronic characteristics that have attracted considerable attention in recent years, of which metal clusters is one of the most essential parts1–15. The fi rst major breakthrough was the observation on “magic numbers” in the mass spectra of free sodium made by Knight and co-workers in 198416, to the extent that much effort was put to understanding their stability in the factor of electronic shell. The first reasonable explanation was on the basis of the spherical jellium model proposed by Clemenger and co-workers17. In this model, the electronic levels of metal clusters are 1S2|1P6|1D10|2S21F14|2P61G18|···, which is associated with the magic numbers 2, 8, 18, 20, 34, 40, 58, ···. Moreover, the fact along with similarity in stability and chemical nature between simple metal clusters and individual atom, the superatom electronic theory has been successful explaining the mass abundances of gas-phase metallic clusters18. Khanna, Jena and Castleman presented that the magic-number metal clusters could be viewed as superatoms19–23.
However, there is no a reasonable explanation to the nonspherical clusters and sub-peaks in the mass spectra by spherical jellium model. In 2013, our group proposed a super valence bond (SVB) model24, of which the shell closures of superatoms were obtained by sharing valence pairs and nuclei with superatoms or ligands. This model has been ideally applied in Li clusters (Li26, Li14, Li10, Li8), Au clusters (Au+293,Au+260, Au20) and other metal clusters24–31.
The chemistry of palladium is one of the most extensive and versatile fields of chemistry, because this metal can easily form complexes with many organic and inorganic molecules32–38. In 2016, Benjamin and co-workers39synthesized an unusual[Pd4(μ3-SbMe3)4(SbMe3)4] cluster which is composed of a tetrahedral Pd(0) core with four terminal SbMe3ligands and four triple bridging SbMe3ligands capping four triangular Pd3faces, inevitably attracting our attention. Theoretical research shows that both bridging and terminal SbMe3 ligands can help to stabilize the electron rich Pd4 core as efficient acceptors in metal-to-ligand π-back-donation. What motivated us to investigate this complex is the unusual geometric shape and complicated electronic structure. We want to see whether the above models can be used to illustrate the stability of this Pd cluster. Our aim is, therefore, to decipher bond nature of the 4-core 8-ligands tetrahedral complex and predict a series of analogues. The understanding of bonding nature between the two kind of ligands and 4-core metal clusters should provide more possibilities to the synthesis of transition metal compounds.
2 Computational methods
The geometries of the studied system in this work are fully optimized at TPSSh/def2-TZVP level. The vibrational frequencies are computed to check whether the obtained structure is a true minimum point with all real frequencies. We also apply the same level to calculate the HOMO-LUMO gaps(EHL), NICS and binding energies. NICS value is a popular measurement for aromaticity, where negative value shows aromaticity, and positive value suggests antiaromaticity.Adaptive natural density partitioning (AdNDP) developed by Boldyrev’s group40is carried out using PBE method with the basis set of LANL2DZ, in order to elucidate the chemical bonding of these clusters. All calculations are done with Gaussian 09 package41. The visualization of the AdNDP results is realized using the MOLEKEL 5.4.0.8 program42.
3 Results and discussion
3.1 Geometry structure
The initial coordinates of [Pd4(μ3-SbMe3)4(SbMe3)4] are obtained from crystal data of the Benjamin paper39. For the convenience of going into the electronic structure of cluster, all methyl groups are replaced with H atoms and optimized at TPSSH/def2-TZVP level (Fig. 1). The ultimate cluster structure is Td[Pd4(μ3-SbH3)4(SbH3)4]. The optimized Pd-Pd distance of Pd4unit and bond length of Pd-SbS/V(bridging (μ3-Sb)/terminal Sb atoms) are 0.2792 nm and 0.2790/0.2537 nm, respectively.Theoretical results are in good agreement with the corresponding experimental values (0.2805 nm and 0.2773/0.2520 nm, respectively). The [Pd4(μ3-SbH3)4(SbH3)4] cluster has an tetrahedron Pd4metal core stabilized by eight ligands. It can be seen that all Pd3faces of tetrahedron are fully capped by SbH3 ligands and all Pd atoms are coordinated to terminal SbH3 ligands.
3.2 Electronic structure
Why is this tetrahedron cluster stable? First, we concentrate on the nature of the bonding in this structure by using AdNDP method which is wildly applied for closed-shell species recovering both the classical Lewis bonding concepts(lone-pairs and two-center two electron (2c-2e) bonds) and the delocalized n-center two electron (nc-2e) bonds43–53.

Fig. 1 Optimized Td structure of [Pd4(μ3-SbH3)4(SbH3)4]cluster at the TPSSh/def2-TZVP level.Pd-green; Sb-brown; H-white.
The Td[Pd4(μ3-SbH3)4(SbH3)4] possesses 104 valence electrons in total. AdNDP analysis (Fig. 2) identifies twenty lone pairs with occupy numbers (ONs) lying within the range of 1.90–2.00 |e|, which are generated by the d orbital electrons of four Pd atoms. There exist clearly 24 Sb―H σ bonds, which consume 48 electrons. This remains 16 electrons, with each Sb atom contributing two valence electrons. 8 electrons are localized along the four terminal Sb―Pd dative bonds. The other 8 electrons are delocalized between four capping Sb atoms and Pd4core. AdNDP analysis gives two bonding patters of the 8 electrons of [Pd4(μ3-SbH3)4(SbH3)4]. Pattern I is based on the SAN model proposed by Cheng et al.24, this 8e cluster should be taken as a network of four 4c-2e tetrahedral PdSb3superatoms. As expected, AdNDP reveals four 4c-2e delocalized super σ-bonds with ON = 1.96 |e| in each PdSb3units, which suggests that [Pd4(μ3-SbH3)4] includes four PdSb3close-shell 2e superatoms, making the [Pd4(μ3-SbH3)4] as an eight electron shell. Pattern II is according to the spherical jellium model, this electron number is consistent with the superatom concept and corresponds to the closed-shell 1S21P6configuration. [Pd4(μ3-SbH3)4] is found with one super S (ON =2.00 |e|) and three P (Px,y,z) (ON = 1.97 |e|) 8c-2e orbitals.

Fig. 2 Bonding pattern for Td [Pd4(μ3-SbH3)4(SbH3)4] cluster as revealed from the AdNDP analysis.Occupation numbers (ONs) are shown.
With the geometric shape and electronic structure of[Pd4(μ3-SbH3)4(SbH3)4], each Pd atom is in 4d10configuration and coordinated to one terminal Sb atom and three bridging Sb atoms, of which sp3empty orbitals are occupied with 8 electrons contributed by four Sb atoms, rendering 18-electrons principle. Meanwhile, [Pd4(μ3-SbH3)4] obtains closed shells by sharing electronic pairs with ligands, being of 8e superatom.
Since the delocalization is an important cause of aromaticity,and aromaticity is usually associated with the high stability, we carry on the study on the aromaticity for this cluster. We believe the aromaticity of Pd4(μ3-SbH3)4is also a key part to govern the stability of Td[Pd4(μ3-SbH3)4(SbH3)4]. Herein,NICS-scan method is employed to analyze the aromaticity of this cluster. The aromatic properties along z-axes (coined as NICSzz-scan) within the range of 0–0.30 nm from center to out-of-plane of Pd4(μ3-SbH3)4, Pd4(SbH3)4and Pd4(μ3-SbH3)4(SbH3)4are calculated. Fig. 3 plots the NICSzz-scan curves for Pd4(μ3-SbH3)4, Pd4(SbH3)4and Pd4(μ3-SbH3)4(SbH3)4. In the case of Pd4(SbH3)4motifs, the NICS values are almost positive, meaning its antiaromaticity.However, all negative NICS values suggest aromaticity of Pd4(μ3-SbH3)4, in contrast to the antiaromaticity of Pd4(SbH3)4.For Td[Pd4(μ3-SbH3)4(SbH3)4] cluster, it also exhibits negative NICS values which are smaller than the that of Pd4(μ3-SbH3)4(within the range of 0 to 0.25 nm). What the consequence can be caused by the antiaromaticity of Pd4(SbH3)4and the aromaticity of Pd4(μ3-SbH3)4 together. NICS calculations agree well with the AdNDP analysis. Moreover, the H-L gap of[Pd4(μ3-SbH3)4(SbH3)4] (2.84 eV) is also very large, indicating the close-shell electronic structure of [Pd4(μ3-SbH3)4(SbH3)4].As predicted, we can draw a conclusion that [Pd4(μ3-SbH3)4(SbH3)4] is stable with high aromaticity, which is based chiefly on Pd4(μ3-SbH3)4.
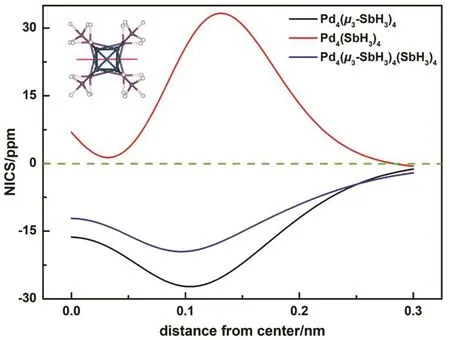
Fig. 3 The NICSzz-scan curves for Pd4(μ3-SbH3)4, Pd4(SbH3)4 and Pd4(μ3-SbH3)4(SbH3)4 within the range of 0 to 0.30 nm from center to out-of-plane.
3.3 M4(μ3-LAH3)4(LBH3)4
The intriguing bonding and aromatocity of Pd4(μ3-SbH3)4(SbH3)4have motivated us to design additional clusters.Here we certify that the new metal clusters can be also 8e superatoms by changing metal and ligand while without changing their valence electron count. Considering the Pd 4d10and Sb 5s25p3configurations, we replace Pd with M (M =4d105s1Ag, 4d105s2Cd, 5s2Ba and 5s25p1In), accordingly, LA and LB (Si, Ge, Sn, Pb and Al) can be substituted for Sb.
To determine the stabilities of the series of analogues, the EHLgaps are calculated and the results are listed in Table 1. By contrast, for M = Ag and Cd, these clusters possess slightly larger EHLvalues from 2.90 to 3.68 eV as a comparison with that of Pd4(μ3-SbH3)4(SbH3)4 of 2.84 eV. Whereas the EHL values of In species are significantly smaller, the reason is probably that, the In is the main group element.
In Table 1, we also calculate the binding energies, which are defined as Eb-S= E[M4(μ3-LAH3)3(LBH3)4] + E[μ3-LAH3] -E[M4(μ3-LAH3)4(LBH3)4], Eb-V= E[M4(μ3-LAH3)4(LBH3)3] +E[LBH3] - E[M4(μ3-LAH3)4(LBH3)3], where E[M4(μ3-LAH3)3(LBH3)4], E[μ3-LAH3], E[M4(μ3-LAH3)4(LBH3)4], E[M4(μ3-LAH3)4(LBH3)3] and E[LBH3] represent the energies of molecules M4(μ3-LAH3)3(LBH3)4, μ3-LAH3, M4(μ3-LAH3)4(LBH3)4, M4(μ3-LAH3)4(LBH3)3and LBH3, respectively. For all investigated clusters, the positive Eb-Sand Eb-Vshow that M4can bind stably with bridging and terminal ligands. In comparison, [Pd4(μ3-SbH3)4(SbH3)4] is most stable with biggest Eb-Sand Eb-Vamong these clusters. We can know that each Ag atom provides one electron to bond with Sb atom, thus, there isno longer enough π-back-donation to keep Ag4core, and the Eb-Vof [Ag4(μ3-SbH3)4(BH3)4], being even less than half of that of [Pd4(μ3-SbH3)4(SbH3)4], so too the other analogues.
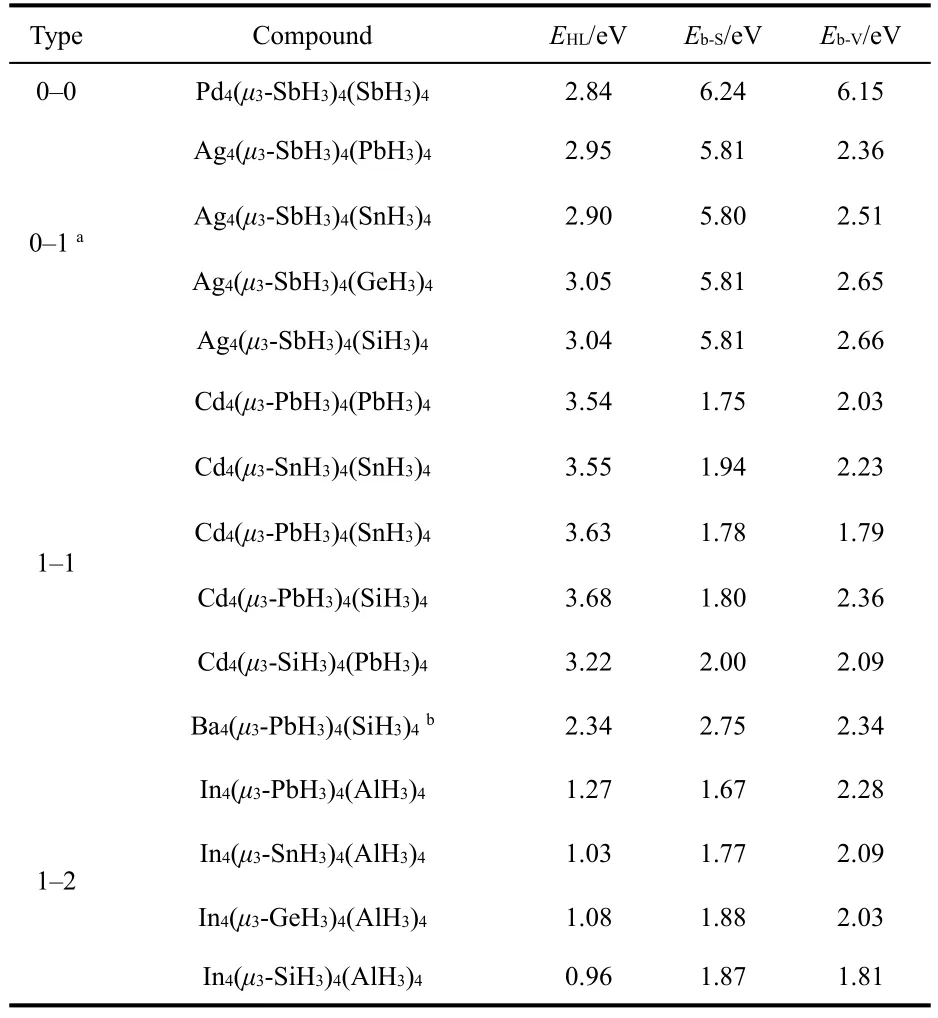
Table 1 HOMO-LUMO gaps EHL of M4(μ3-LAH3)4(LBH3)4, binding energies Eb-S, Eb-V of M4(μ3-LAH3)4 and M4(LBH3)4, respectively.

Fig. 4 Bonding pattern for Td [Ag4(μ3-SbH3)4(SiH3)4] cluster as revealed from the AdNDP analysis.Occupation numbers (ONs) are shown.
The chemical bonding analyses of the series of analogues are also given by AdNDP method. One example is [Ag4(μ3-SbH3)4(SiH3)4], which has the same valence electrons with[Pd4(μ3-SbH3)4(SbH3)4] (Ag 4d105s1versus Pd 4d10, Si 3s23p2versus Sb 5s25p3). The [Ag4(μ3-SbH3)4(SiH3)4] (Fig. 4) features a similar bonding pattern of [Pd4(μ3-SbH3)4(SbH3)4], with twenty lone pairs (dxy, yz, xz, x2- y2, z2), twelve Si―H σ bonds, twelve Sb―H σ bonds, four terminal Ag―Si σ bonds and four 4c–2e super σ-bonds or four 8c–2e super S and Px,y,zorbitals.
3.4 Closed-shell interaction
Note that the calculated Pd-Pd distance is 0.2792 nm, which turns out to be longer than a single bond (0.2400 nm)54but remarkably smaller than the van der Waals radius. The four Pd atoms are in Pd(0) configuration, the interaction in Pd4tetrahedron that is similar to aurophilicity in Au-Au clusters55–59. It is also found that there are large difference of stability between transition metal clusters and non-transition metal clusters.

Fig. 5 BSSE-corrected dissociation energy curves of M–M (M = Ag, K) interaction.
For all of the reasons mentioned above, to verify the existence of closed-shell interaction in this Pd-Pd cluster, we futher design two model clusters, Td Ag4Cl4 and Td K4Cl4. The structures of two model clusters are remarkably similar with respect to Pd4(SbH3)4motifs in Pd4(μ3-SbH3)4(SbH3)4, despite the imaginary frequencies they have. Curves for the M–M (M =Ag, K) interaction energy (Eint= 4E[MCl] - E[M4Cl4]) of two model clusters as the M-M distance are illustrated in Fig. 5.There is one trough of Eintcurve in Ag4Cl4cluster, where the Eintvalues are negative, indicating the attractive interaction between Ag atoms, however, K4Cl4is not. This is because the former has d10valence electrons but the latter does not. It is necessary to point out that d10···d10interaction can stabilize the pd4 tetrahedron structure.
4 Concliusions
In summary, we present the structure and chemical bonding of Td[Pd4(μ3-SbH3)4(SbH3)4] and the series of analogues by the computational study. Each Pd atom in Pd4(0) tetrahedron is coordinated to one terminal Sb atom and three bridging Sb atoms with sp3hybridization, fulfilling with 4d, 5s and 5p orbitals, rendering 18-electrons principle. In terms of[Pd4(μ3-SbH3)4] motifs, it can be not only viewed as a network of four 4c–2e superatoms capping on Pd4core or a 8e shell with four 4c–2e super σ-bonds based on SAN and SVB model,but also as a 8e-superatom with 1S21P6jellium closed-shell configuration. At the same time, the d10···d10interaction plays a critical role in stabilizing the Pd4tetrahedron structure. The[Pd4(μ3-SbH3)4(SbH3)4] is demonstrated to be stable with a filled electronic shell, a large HOMO-LUMO gap and negative NICS value, and the NICSzz-scan suggests that the aromaticity of [Pd4(μ3-SbH3)4(SbH3)4] can be derived from Pd4(μ3-SbH3)4.Moreover, the calculations of energies and bonging analysis of AdNDP reveal that these designed Ag and Cd analogues are also stable structures though they are not better than[Pd4(μ3-SbH3)4(SbH3)4], but still being significant in designing and synthesizing such metal clusters.
Acknowledgment: The calculations were carried out at the High-Performance Computing Center of Anhui University.
杂志排行

物理化学学报的其它文章
- Synthesis of High Yield Au21(SR)15 Nanoclusters
- Luminescence Emission of Copper Nanoclusters by Ethanol-induced Aggregation and Aluminum Ion-induced Aggregation
- Construction and NIR Luminescence Properties of Zn-Ln Rectangular Nanoclusters
- Ag7(MBISA)6 Nanoclusters Conjugated with Quinacrine for FRETEnhanced Photodynamic Activity under Visible Light Irradiation
- Synthesis and Structure Determination of Ag-Ni Alloy Nanocluster Ag4Ni2(SPhMe2)8 (SPhMe2 = 2,4-dimethylbenzenethiol)
- PPh3: Converts Thiolated Gold Nanoparticles to [Au25(PPh3)10(SR)5Cl2]2+