A Preliminary Genetic Linkage Map of Sinonovacula constricta(Lamarck, 1818) Based on Microsatellites Derived fromRAD Sequencing
2018-08-24WUXuepingFENGYanweiJIANGHailinLIUXiangquanandPANYing
WU Xueping, FENG Yanwei, JIANG Hailin, LIU Xiangquan, *,and PAN Ying
A Preliminary Genetic Linkage Map of(Lamarck, 1818) Based on Microsatellites Derived fromRAD Sequencing
WU Xueping1), 2), 3), #, FENG Yanwei1), #, JIANG Hailin1), LIU Xiangquan1), *,and PAN Ying2), *
1),,264006,2),,530004,3),,530008,
isone of the important economic aquaculture species in China. In this study, we constructed genetic linkage maps ofbased on 300 microsatellite markers derived from RAD-seq using an F1 full-sib family. The female map contained 204 microsatellites assigned to 22 linkage groups, which covered 1529.5cM with an average interval of 10.3cM. The male consisted of 187 microsatellites in 19 linkage groups corresponding to the haploid chromosome number (n=19), which spanned 1429.3cM with an average interval of 8.7cM. The genome coverage was approximately 83.5% and 81.4%, respectively. An integrated map was constructed according to the common markers in parental linkage groups, which had a total length of 1683.8cM with an average interval of 7.3cM. The genome coverage of the integrated map was approximately 86.3%. The genetic linkage map would form the foundation for further studies on the quantitative trait loci (QTL), as well as accelerating the breeding process of this species.
linkage maps;; microsatellite marker; RAD sequencing
1 Introduction
Razor clam,, belonging to Mollusca (Bivalvia: Veneroida: Solecurtidae), is widely distributed in the intertidal coastal and estuarine waters of China, Japan, Korea and Vietnam (Xu and Zhang, 2008). It is one of the four major commercial aquatic clams in China.As well as,and,has been farmed for over 500 years in Zhejiang and Fujian Provinces due to its short culture cycle, high nutritive value and delicious taste (Xie, 2003). In recent years, culturinghas expanded greatly, and the output has reached 793708 tons (FAO, 2016). However, there are many challenges in the artificial culture process, such as low growth rate, disease and low survival (Feng., 2010). To counter these challenges, conventional selections have been performed (Du., 2016; Li., 2016). However, directional selections that depend onphenotype are time-consuming, labor-intensive and ex- pensive (Guo., 2012).
Generally, the economic traits of a species, such as rapid growth and disease resistance, are determined by multiple loci and are quantitatively inherited. Molecular markers provide a powerful tool for identification of quantitative trait loci (QTL) in a selective program. In contrast to traditional selection, marker-assisted selection (MAS) and phenotypic selection in breeding strategies can greatly accelerate breeding efforts. A saturated gene- tic linkage map is regarded as an essential step towards establishing MAS programs and allowing QTL detection. Genetic maps have been constructed in many important aquaculture species,..,(Zhan.,2012),(Liu., 2013),(Luzón., 2015), and(Qiu., 2016).
Construction of a fine genetic map requires many mo- lecular markers. Microsatellites (or simple sequence re- peats, SSR) are easily transferrable among laboratories and are ideal co-dominant marker systems (Song., 2012; Chu., 2014; Tsigenopoulos., 2014). The development of microsatellite markers using traditional methods requires significant efforts. In contrast, restric- tion site-associated DNA sequencing (RAD-seq), a next- generation sequencing (NGS) technology, provides a more cost-effective and higher throughput method to generate useful markers and has been widely used to study non- model organisms (Baird., 2008; Gonen., 2014).
In this study, we constructed genetic linkage maps ofbased on microsatellite markers derived from RAD-seq, aiming at providing the basis for future QTL- mapping and MAS efforts.
2 Materials and Methods
2.1 Mapping Family and DNA Extraction
According to the pseudo-testcross mapping strategy, an F1 full-sib family was established by single-pair mating a wild female and a wild male in October 2012, which were both captured from Rizhao, Shandong Province, China. After 10-day rearing, 150 early umbo-veliger larvae were randomly selected and kept in 100% ethanol (changed three times). Adductor muscle tissues of the parents were also sampled. All samples were frozen at −20℃ until DNA extraction.
Parental DNA was extracted with the standard phenol- chloroform method (Sambrook and Russell, 2000). Larval DNA was prepared using the Chelex-100 extraction tech- nique as previously described (Li and Kijima, 2006), fol- lowed by amplifying genomic DNA with REPLI-g®Mini Kit(QIAGEN, Duesseldorf, Germany).
2.2 RAD Library Preparation and Sequencing
DNA sample was obtained from the adductor muscle of a living(named SC15, sampled from Yantai, Shandong Province, China), using the same method as the parental DNA. Genomic DNA digested byI and an adapter (P1) was ligated to the fragment’s compatible ends. This adapter contained forward amplification and Illumina sequencing primer sites, as well as a nucleotide barcode for sample identification. The adapter-ligated fragments were subsequently pooled, randomly sheared and ligated to a second adapter (P2) that has divergent ends. Finally, the fragments of 200bp to 400bp were col- lected for libraries construction. After quality assessment, the libraries were sequenced on an Illumina HiSeq2500platform and 125bp paired-end reads were generated.
2.3 RAD-seq Data Assembly and MicrosatelliteDevelopment
Prior to the assembly, stringent filtering was conducted, and only high-quality reads were sent to the VelvetOpti- miser assembler with parameter settings: -s 23 -e 31 -x 4 (Daniel., 2008). The contigs assembled were as ref- erence genome for obtaining double-end SSR (100bp) fragments. SSR primers were designed using PRIMER 3.0 program (http://www.premierbiosoft.com/).
2.4 Microsatellite Analysis
Markers were genotyped in the parents and six progeny to look for polymorphism. PCR was amplified in 10µL reactions containing 0.25UDNA polymerase (Ta- kara, Dalian, China), 1× PCR buffer (Mg2+), 0.2mmolL−1dNTP mix, 0.5µmolL−1of each primer set. PCR program (Li., 2016) performed as the following thermal cy- cles: 3min at 94℃; seven cycles of 1min at 94℃, 30s at the optimal annealing temperature, and 30s at 72℃; 33 cycles of 30s at 94℃, 30s at the specific annealing tem- perature, 30s at 72℃, and a final extension of 5min at 72℃. The PCR products were evaluated by electrophore- sis on 8% non-denaturing polyacrylamide gel and visual- ized by silver staining.
Polymorphic microsatellites were used in subsequent genotyping of parents and 150 offspring to construct the linkage maps. Forward primer was labeled with either FAM or HEX fluorescence dye in Sangon BioTechno- logies, Shaihai, China. PCR reactions were carried out in 25µL volumes: 1μmolL−1of each primer set, 12.5µL 2× Easy Taq mix (Transgen, Beijing, China), and about 50ng template DNA. The PCR conditions were as follows: an initial denaturation for 3min at 94℃, followed by 35 cycles 45s at 94℃, 45s at primer-specific annealing temperature, 3min at 72℃, with a final extension of 5min at 72℃. Product of one locus was added with 0.1µL GeneScanTM-500 LIZTMsize standard (ABI, USA) and 9.9µL Hi DiTMformammide, and subjected to genotyping on an ABI 3700XL. Data collection was performed on GeneMapper ver. 3.7 software (Applied Biosystems, Fors- ter City, CA, USA).
2.5 Segregation and Map Construction
A linkage map was constructed using JoinMap 4.0 soft- ware (van Ooijen, 2006) under cross-pollinating (CP) coding scheme, which handles F1 outbred population data containing genotype configurations with unknown lin- kage phase. Basing on JoinMap software, segregation distortion of markers was detected by a chi-square (χ2) with a level of<0.01. Corrections of the significance level for multiple tests were performed following the se- quential Bonferroni correction. In order to avoid false linkages, markers deviating from Mendelian expectations after Bonferroni correction were excluded from the link- age analysis. Linkage groups were determined at an in- dependence LOD (logarithm of odds) threshold of 4.0 and a recombination threshold of 0.40. Recombination fre- quencies were converted into genetic distances using the Kosambi mapping function (Kosambi, 1944). Once the female and male maps were established, a consensus map was obtained using the ‘Map Integration’ function. The linkage map was drawn by MapChart 2.3 software (Voor- rips, 2002).
2.6 Genome Size and Coverage
The observed genome length (Go) was estimated by adding the length of each linkage group. The expected genome length (Ge) was calculated using two methods: (i)e1=of+2(Fishmen., 2001), whereofis the length of each linkage group excluding triplets and dou- blets, whilerepresents the average spacing between markers, denoted by dividing the total observed length of all linkage groups by the number of intervals (number of markers minus the number of linkage groups); (ii)e2was calculated by multiplying the length of each linkage group by a factor of (+1)/(−1), whereis the num- ber of markers of each linkage group (Chakravarti., 1991). The average ofe1ande2was used ase. The map coverage was calculated byo/e.
3 Results
3.1 RAD Sequencing Analysis
After stringent quality assessment and data filtering, a total of 48810439 clean reads were obtained, which were assembled into 498746 contigs. Totally 5700 pairs of mi- crosatellite primers were designed for subsequent analysis (Table 1).

Table 1 Summary of sequencing data
3.2 Markers Analysis and Segregation Distortion
A total of 656 primer pairs were selected to test ampli- fication conditions and polymorphism. The result showed that 300 markers (45.7%) exhibited heterozygosity in at least one parent. Of these 300 markers, 127 markers were polymorphic in both parents and could be used to integrate the map, 99 markers were polymorphic only in female parent, and 74 markers were polymorphic only in male parent. Thus, 226 markers were used to construct the female map, and 201 markers were used to construct the male map.
Chi-square analysis indicated that 19 markers deviated from Mendelian expectation at<0.01 after Bonferroni correction among the 226 female markers. For the 201 male markers, 11 markers exhibited significant deviation from Mendelian expectation.
3.3 Linkage Analysis and Map Coverage
The final length of the female map contained 22 linkage groups with 204 markers, covering 1529.5cM (Fig.1 and Table 2). Lengths of linkage groups varied between 17.8cM and 138.5cM. The resolution ranged from 4.8cM to 30.7cM, with an average of 10.3cM. The marker den- sity was 2-26 per group. The expected genome length (Ge) was 1831.2cM. The genome coverage of the female map was approximately 83.5% (Table 3).

Fig.1(a)Genetic linkage maps ofFemale map (F) and male map (M) are shown on the left and right, respectively. The integrated genetic map is shown on the center. Map distance is given in cM at the left of each linkage group (LG), while marker names are given on the right. The female map was matched to the male map using common markers (F1/M1, F2/M3).
Fig.1(b)Same as Fig.1(a) but the female map using common markers (F3/M13, F4/M8).

Fig.1 (c)Same as Fig.1(a) but the female map using common markers (F5/M9, F6/M12).

Fig.1(d)Same as Fig.1(a) but the female map using common markers (F7/M14, F8/M17).
Fig.1(e)Same as Fig.1(a) but the female map using common markers (F9/M5, F10/M7).
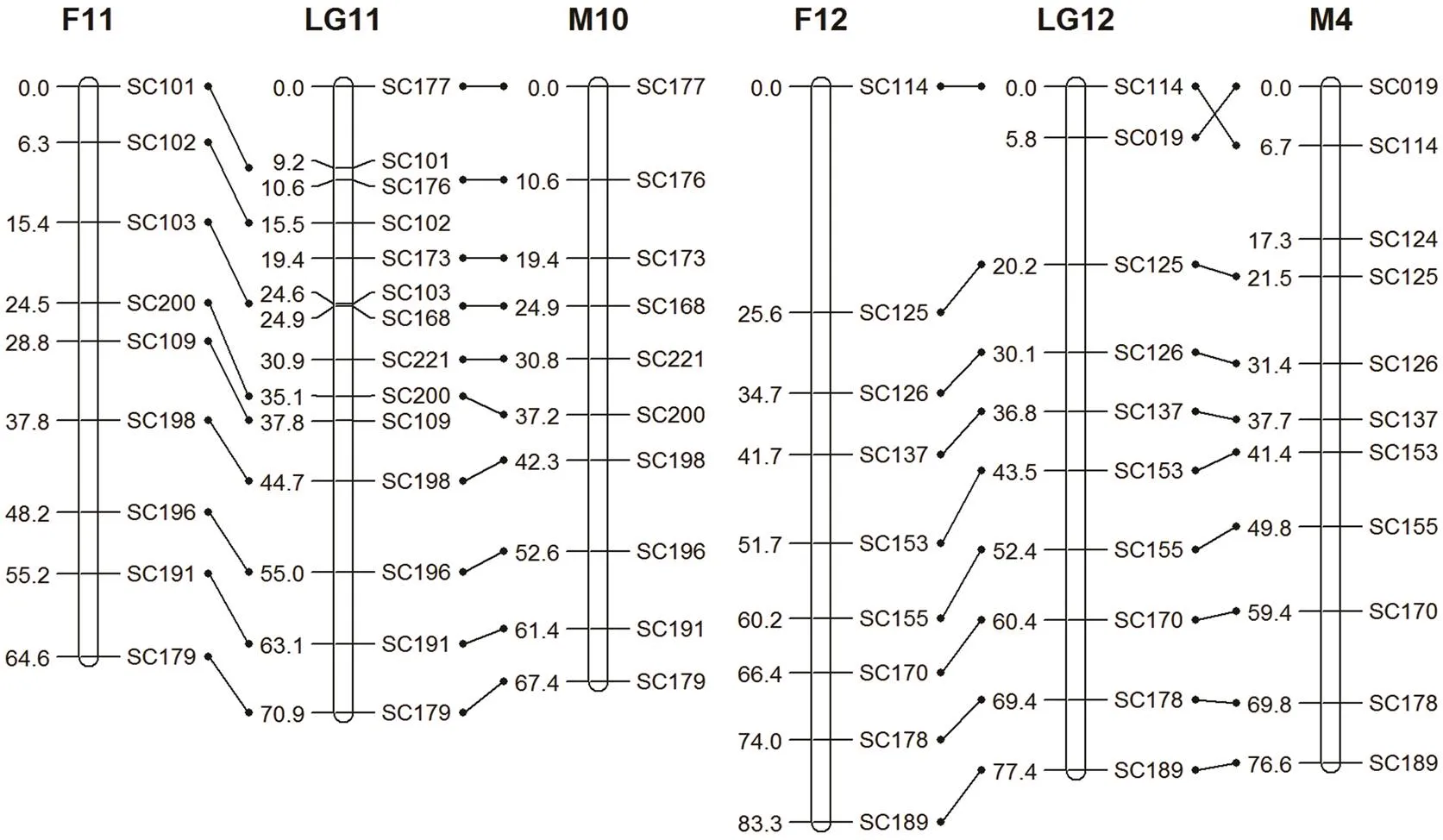
Fig.1(f)Same as Fig.1(a) but the female map using common markers (F11/M10, F12/M4).
Fig.1(g)Same as Fig.1(a) but the female map using common markers (F15/M2, F14/M15).
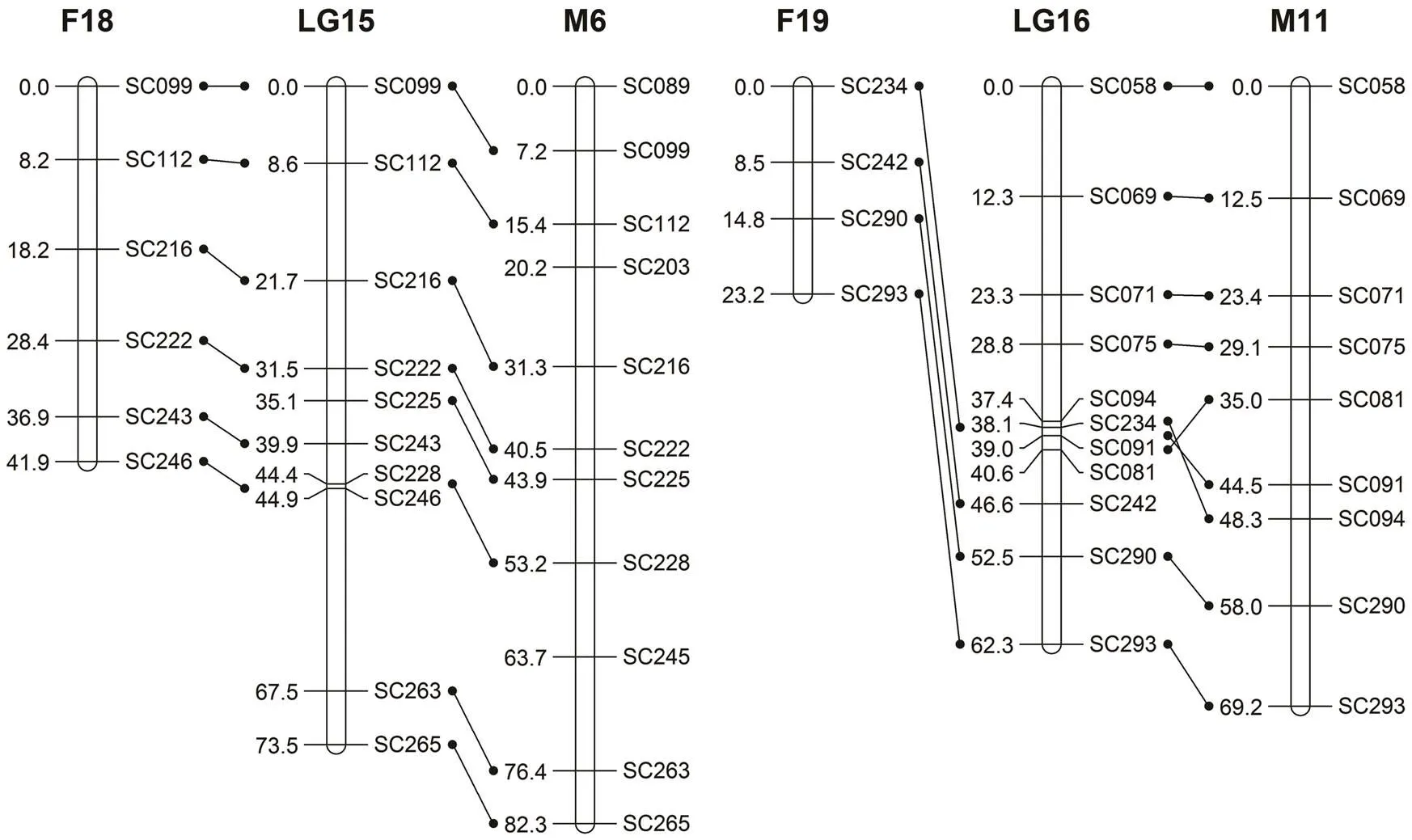
Fig.1(h)Same as Fig.1(a) but the female map using common markers (F18/M6, F19/M11).
Fig.1(i)Same as Fig.1(a) but the female map using common markers (F20/M16, F21/M19).

Fig.1(j)Same as Fig.1(a) but the female map using common markers (F22/M18, F13, F16, F17).
Table 2 Information of the genetic linkage maps of


Table 3 Summary of the genetic linkage maps of S. constricta
The male map consisted of 19 linkage groups with 187 markers, spanning a total genetic distance of 1429.3cM (Fig.1 and Table 2). The length of each linkage group ranged from 27.0cM to 152.1cM and contained 4-20 markers. The interval of markers ranged from 7.4cM to 14.9cM, with a mean of 8.7cM. The expected genome length was 1756.3cM. The genome coverage was about 81.4% (Table 3).
The female map was matched to the male map using common markers (F1/M1, F2/M3, F3/M13, F4/M8, F5/ M9, F6/M12, F7/M14, F8/M17, F9/M5, F10/M7, F11/M10, F12/M4, F15/M2, F14/M15, F18/M6, F19/M11, F20/ M16, F21/M19, and F22/M18). The integrated map mea- sured 1683.8cM with a total of 270 markers distributed over 19 linkage groups. The length of each group ranged from 25.6cM to 186.0cM. The minimum and maximum intervals were 4.6cM and 11.4cM, respectively. The expected genome length was 1950.2cM. The genome coverage was approximately 86.3%, higher than those of the female and male maps. Linkage group 1 (LG1) included the most markers (Fig.1, Table 2 and Table 3).
3.4 Differences of Recombination Between Sexes
Compared with the male map (1429.3cM), the female map (1529.5cM) showed significantly higher recombi- nation ratio. For the integrated map, the length of some groups (LG2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14) were longer in the female map than in the male map while that of other groups were longer in the male map than in the female map (LG1, 11, 13, 15, 16, 17, 18, 19). The total length of the intervals between common markers for the female and male map was 197.7cM and 165.5cM, respectively. The female-to-male recombination ratio was 1.19:1.
4 Discussion
4.1 RAD-seq Technology
Compared to the whole-genome shotgun (WGS) sequencing, RAD-seq technology is a powerful method to analyze genomic resources in non-model organisms lack- ing a reference genome, and has been successfully used for SSR or SNP discovery and genotyping. This technique provides a great opportunity to overcoming the limita- tions of traditional methods (da Fonseca., 2016). In this study, a total of 498746 contigs were assembled and 5700 pairs of primers were obtained, much more than those reported in previous studies (Niu., 2008; Jiang., 2010; Ma., 2015; Wu., 2015), which demonstrated that RAD-seq is a cost-effective and high- throughput method for the discovery of microsatellites in.
4.2 Segregation Distortion
The observed distortion rate was 8.5% for the female parent and 5.6% for the male parent. Compared to other aquatic animals, this percentage was 18.8% less than EST-SSRs and SSRs in the Pacific oyster (Guo., 2012), 19.3% of SSRs in the scallop(Li., 2012), 16.8% (female) and 15.3% (male) of SSRs in the Chinese mitten crab(Qiu., 2016). It was similar to 6.0% of SSRs in tilapia (Lee., 2005). The causes of this apparent variation of distortion rate have not been determined. Preferential fertilization (Lyttle, 1991), high genetic load, zygotic se- lection (Liu., 2010), scoring errors, amplification of the same size from different genomic regions (Fairs., 1998), and non-random sampling or sampling in finite populations (Rick, 1969) may contribute to the segrega- tion distortion.
4.3 Linkage Analysis
In this study, we constructed the first genetic linkage map using 300 SSR markers and genotyped 150 F1 offspring from a full-sib family. After excluding the sig- nificant segregation distortion (<0.01), the linkage map was constructed at a threshold of a log of odds (LOD) score of 4.0. The remaining 204 markers were assigned to 22 linkage groups in the female map, and 187 markers were arranged into 19 linkage groups in the male map. The integrated map contained 19 linkage groups. The number of linkage groups in male map and integrated map were consistent with the haploid chromosome num- ber (n=19) of(Wang., 1998). The re- sults indicated that the selected markers did not distribute evenly and more markers are required to adequately cover the linkage groups and obtain full genome coverage.
The observed map length was 1529.5cM and the ge- nome coverage was 83.5% for female, and 1429.3cM and 81.4%, respectively for male. The integrated map covered 1683.8cM with map coverage being 86.3%. These values were lower than those of other mollusks linkage maps like those for(Bai., 2015) and(Shi., 2014), but larger than(Zhan., 2009) and(Petersen., 2012)This may be due to variable marker densities However, the female map had three more linkage groups than the male map and loca- tions of loci between some linkage groups (for example, F1 and M1). Additionally, some female linkage groups, F13, F16 and F17 were not integrated. These discrepan- cies might be due to low density and small sample size, different markers between the female map (204) and the male map (187), and uneven distribution of markers. A similar phenomenon was reported in other species, such as Pacific oyster(Zhong., 2014), large yellow croaker(Ye., 2014), half smooth tongue sole(Jiang., 2013), and turbot(Ruan., 2010).
4.4 Recombination Rate Between Sexes
The recombination rate in female is commonly higher than in male in many species, such as(1.67:1, Sahoo., 2015), red drum (1.14:1, Hollenbeck., 2015), black tiger shrimp (1.60:1, Baranski., 2014), and grass carp (1:(2.0–2.03), Xia., 2010). However, in our study, some male linkage groups showed a higher recombination rate than female. There is dis- agreement among scholars about the explanation of the differences in the recombination rate. One hypothesis suggested that sex differences might account for more suppressed recombination rates in the heterogametic sex than homogametic sex (Haldane, 1922; Huxley, 1928; Singer., 2002), but Cui. (2015) and Liu. (2013) proposed that sexual environment in embryologi- cal stage plays a more important role in determination of recombination rates than genetic sex. Many other factors, including sexual selection (Triver, 1988), meiosis (Lin- dahal, 1991), and chromosome structure (Lynn., 2004), might explain recombination rates. Further inves- tigation is needed to understand the mechanism causing sex differences in recombination rate.
5 Conclusions
In this study, we developed a large number of microsatellites with RAD-seq technology and constructed genetic linkage maps ofusing 300 polymorphic microsatellite markers. The maps ofcan provide the framework to identify QTL and facilitate future selective breeding programs in this important resource.
Acknowledgements
The study was supported by the grants from the Natural Science Foundation of Shandong Province (No. ZR2012 CM037), the Shandong Provincial Agriculture Thorough- bred Project, and the Innovation Project of Guangxi Graduate Education (No. YCBZ2015007).
Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A., Seiker, E. U., Cresko, W. A., and Johnson, E. A., 2008. Rapid SNP discovery and genetic mapping using sequencing RAD markers., 3: e3376.
Bai, Z. Y., Han, X. K., Luo, M., Lin, J. Y., Wang, G. L., and Li, J. L., 2015. Constructing a microsatellite-based linkage map and identifying QTL for pearl quality traits in triangle pearl mussel ()., 37: 102-110.
Baranski, M., Gopikrshna, G., Robinson, N. A., Katneti, V. K., Shekhar, M. S., Shanmugakarthik, J., Jothivel, S., Gopal, C., Ravichandran, P., and Kent, M., 2014. The development of a high density linkage map for black tiger shrimp () based on cSNPs., 9: e85413.
Cervera, M. T., Storme, V., Ivens, B., Gusmão, J., Liu, B. H., Hpstyn, V., Slycken, J. V., Montagu, M. V., and Boerjan, W., 2001. Dense genetic linkage maps of threespecies (,and) based on AFLP and microsatellite markers., 158: 787-809.
Chakravarti, A., Lasher, L. K., and Reefer, J. E., 1991. A maximum likelihood method for estimating genome length using genetic linkage data., 128: 175-182.
Chu, G. N., Jiang, L. M., Yan, H., Yu, H. Y., Wang, Z. G., Jiang, H. B., and Zhang, Q. Q., 2014. A micirosatellite genetic linkage map of black rockfish ()., 13: 1078-1086.
Cui, Z., Hui, M., Liu, Y., Song, C., Li, X., Li, Y., Liu, L., Shi, G., Wang, S., Li, F., Zhang, X., Liu, C., Xiang, J., and Chu, K. H., 2015. High-density linkage mapping aided by transcrip- tomics documents ZW sex determination system in the Chinese mitten crab., 115: 206-215.
da Fonseca, R. R., Albrechtsen, A., Themudo, G. E., Ramos- Madrigal, J., Sibbesen, J. A., Maretty, L., Zepeda-Mendoza, M. L., Campos, P. F., Heller, R., and Pereira, R. J., 2016. Next-generation biology: Sequencing and data analysis approached for non-model organisms., 30: 3- 13.
Daniel, R. Z., and Ewan, B., 2008. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs., 18: 821-829.
Du, W. J., Wang, C. D., Wang, J., Li, L. X., Niu, D. H., Li, J. L., and Shen, H. D., 2016. Comparison of early growth inselection group and family.,40: 604-611 (in Chinese with English abstract).
Fais, J., Ladomada, B., and Gill, B., 1998. Molecular mapping of segregation distortion loci in., 149: 319-327.
Feng, B., Dong, L. L., Niu, D. H., Meng, S., Zhang, B., Liu, D. B., Hu, S. N., and Li, J. L., 2010. Identification of immune genes of the agamaki clam () by sequencing and bioinformatic analysis of ESTs., 12: 282-291.
Fishman, L., Kelly, A. J., Morgan, E., and Willis, J. H., 2001. A genetic map in thespecies complex reveals transmission ratio distortion due to heterospecific interactions.,159: 1701-1716.
Franch, R., Louro, B., Tsalavouta, M., Chatziplis, D., Tsigeno- poulos, C. S., Sarropoulou, E., Antonello, J., Magoulas, A., Mylonas, C. C., Babbucci, M., Patarnello, T., Power, D. M., Kotoulas, G., and Bargelloni, L., 2006. A genetic linkage map of the hermaphrodite teleost fishL., 174: 851-861.
Gonen, S., Lowe, N. R., Cezard, T., Gharbi, K., Bishop, S., and Houston, R., 2014. Linkage maps of the Atlantic salmon () genome derived from RAD sequencing., 15: 166.
Guo, X., Li, Q., Wang, Q. Z., and Kong, L. F., 2012. Genetic mapping and QTL analysis of growth-related traits in the Pacific oyster., 14: 218-226.
Haldane, J. B. S., 1922. Sex ration and unisexual sterility in hybrid animals., 12: 101-109.
Hollenbeck, C. M., Portnpy, D. S., and Gold, J. R., 2015. A genetic linkage map of red drum () and comparison of chromosomal syntenies with four other fish species., 435: 265-274.
Huxley, J. S., 1928. Sexual difference of linkage in., 20: 145-156.
Ihara, N., Takasuga, A., Mizoshita, K., Takeda, H., Sugimoto, M., Mizoguchi, Y., Hirano, T., Itoch, T., Watanabe, T., Reed, K. M., Snelling, W. M., Kappes, S. M., Beattie, C. W., Beatti, C. W., Benett, C. L., and Sugimoto, Y., 2004. A comprehen- sive genetic map of the cattle genome based on 3802 micro- satellites., 14: 1987-1998.
Jiang, L. M., Chu, G. N., Zhang, Q. Q., Wang, Z. G., Wang, X. B., Zhai, J. M., and Yu, H. Y., 2013. A microsatellite genetic linkage map of half smooth tongue sole ()., 9: 17-23.
Jiang, Q., Li, Q., Yuan, Y., and Kong, L. F., 2010. Polymorphic microsatellite loci for population studies of the razor clam,., 2: 81-83.
Kosambi, D. D., 1994. The estimation of map distances from recombination values., 12: 172-175.
Lee, B. Y., Lee, W. J., Streelman, J. T., Carleton, K. L., Howe, A. E., Hulata, G., Slettan, A., Stern, J. E., Terai, Y., and Ko- cher, T. D., 2005. A second generation genetic linkage map of tilapia (spp)., 170: 237-244.
Li, L. X., Du, W. J., Wang, C. D., Wang, J., Niu, D. H., Li, J. L., and Shen, H. D., 2016. Comparative analysis of growth and heat tolerance, salt tolerance traits amongfamilies., 25: 516-521 (in Chinese with English abstract).
Li, H. J., Liu, X., and Zhang, G. F., 2012. A consensus microsatellite-based linkage map for the hermaphroditic bay scallop () and its application in size-re- lated QTL analysis., 7: e46926.
Li, Q., and Kijima, A., 2006. Microsatellites analysis of gyno- genetic families in the Pacific oyster,., 331: 1-8.
Li, Q., Qi, M. J., Nie, H. T., Kong, L. F., and Yu, H., 2016. Microsatellite-centromere mapping in Japanese scallop () through half-tetrad analysis in gyno- genetic diploid families., 15: 541-548.
Lindahal, K. F., 1991. His and hers recombinational hotspots., 7: 273-276.
Liu, B., Wang, Q. Y., Li, J., and He, Y., 2010. A genetic linkage map of marine shrimp()based on AFLP, SSR and RAPD markers., 28: 815-825.
Liu, F., Sun, F., Li, J., Xia, H. J., Lin, G., Tu, R. J., and Yue, H. G., 2013. A microsatellite-based linkage map of salt tolerant tilapia (×spp.) and mapping of sex-determining loci., 14: 58-71.
Liu, Q., Sakamoto, T., Kubota, S., Okamoto, N., Yamashita, H., Takagi, M., Shigenobu, Y., Nakamura, Y., Sano, M., Wuthi- suthimethavee, S., and Ozaki, A., 2013. A genetic linkage map of kelp grouper () based on microsatellite markers., 415: 63-81.
Luzón, M. J. M., Hermida, M., Pérez, N. R., Robles, F., Navas, J. I., Rejón, C. R., Bouza, C., Martínez, P., and de la Herrán, R., 2015. First haploid genetic map based on microsatellite markers in Senegalese sole (, Kaup 1858)., 17: 8-22.
Lyttle, T. W., 1991. Segregation distorters., 25: 51-557.
Lynn, A., Ashley, T., and Hassold, T., 2004. Variation in human meiotic recombination., 5: 317-349.
Ma, H. T., Jiang, H. B., Liu, X. Q., Wu, X. P., and Wei, X. M., 2015. Polymorphic microsatellite loci for the razor clam,., 14 (1): 145-148.
Niu, D. H., Li, J. L., and Liu, D. B., 2008. Polymorphic microsatellite loci for population studies of the razor calm,., 9: 1393- 1394.
Petersen, J. L., Baerwald, M. R., Ibarra, A. M., and May, B., 2012. A first-generation linkage map of the Pacific lion-paw scallop ()., 350: 200-209.
Qiu, G. F., Xiong, L. W., Liu, Z. Q., Yan, Y. L., and Shen, H., 2016. A first generation microsatellite-based linkage map of the Chinese mitten craband its application in quantitative trait loci (QTL) detection., 451: 223-231.
Rick, C. M., 1969. Controlled introgression of chromosomes ofinto: Segregation and recombination., 62: 753.
Ruan, X. H., Wang, W. J., Kong, J., Yu, F., and Huang, X. Q., 2010. Genetic linkage mapping of turbot (L) using microsatellite markers and its application in QTL analysis., 308: 899-100.
Sahoo, L., Patel, A., Sahu, B. P., Mitra, S., Meher, P. K., Mahapatra, K. D., Dash, K. S., Jayasankar, P., and Das, P., 2015. Preliminary genetic linkage map of Indian major carp,(Hamilton 1822) based on microsatellite markers., 94: 271-277.
Sambrook, J. D., and Russell, W., 2000.. 2nd edition. Cold Spring Harbor Labora- tory Press, Cold Spring Harbor, New York, 543-554.
Shi, Y. H., Wang, S., Gu, Z. F., Lv, J., Zhan, X., Yu, C. C., Bao, Z. M., and Wang, A. M., 2014. High-density single nucleo- tide polymorphism linkage and quantitative trait locus mapping of the pearl oyster,Dunker., 434: 376-384.
Singer, A., Perlman, H., Yan, Y., Walker, C., Corlry-Smith, G., Brandhorst, B., and Postlethwait, J., 2002. Sex-specific re- combination rates in zebrafish ()., 160: 649-657.
Song, W., Li, Y., Zhao, Y., Yan, Y., Liu, Y., Niu, Y., Pang, R., Miao, G., Liao, X., Shao, C., and Gao, F., 2012. Construction of a high-density microsatellite genetic linkage map and mapping of sexual and growth-related traits in half-smooths tongue sole ()., 7: e52097.
Trivers, R., 1988. Sex differences in rated of recombination and sexual selection. In:. Michod, R. E., and Levin, B. R., eds., Sinauer Associates, Sunderland MA, 270- 286.
Tsigenopoulos, C. S., Louro, B., Chatziplis, D., Lagnel, J., Vogiatzi, E., Loukovitis, D., Franch, R., Sarropoulou, E., and Power, D. M., 2014. Second generation genetic linkage map for the gilthead sea breamL., 18: 77-82.
van Ooijen, J. W., 2006. JoinMap®4, Software for calculation of genetic linkage maps in experimental populations. Wage- ningen, Netherlands.
Voorrips, R. E., 2002. MapChart: Software for the graphical presentation of linkage maps and QTLs., 93: 77-78.
Wang, J. X., Zhao, X. F., Zhou, L. H., and Xiang, J. H., 1998. Research on chromosomes of., 29: 191-196 (in Chinese).
Wu, X. P., Feng, Y. W., Ma, H. T., Pan, Y., and Liu, X. Q., 2015. Characterization of new microsatellite loci from the razor clam () and transferability to related species., 61: 175- 178.
Xia, J. H., Liu, F., Zhu, Z. Y., Fu, J., Feng, J., Li, J., and Yue, G. H., 2010. A consensus linkage map of the grass carp () based on microsatellites and SNPs., 11: 135.
Xie, Z. M., 2003.. China Agriculture Press, Beijing, 23-27.
Xu, F. S., and Zhang, S. P., 2008.. Science Press, Beijing, 211-213 (in Chinese).
Ye, H., Liu, Y., Liu, X. D., Wang, X. Q., and Wang, Z. Y., 2014. Genetic mapping and QTL analysis of growth traits in the large yellow croaker., 16: 729-738.
Yu, Z. N., and Guo, X. M., 2003. Genetic linkage map of the eastern oysterGmelin., 204: 327-338.
Zhan, A., Hu, J., Hu, X., Hui, M., Wang, M., Peng, W., Huang, X., Wang, S., Lu, W., Sun, C., and Bao, Z., 2009. Construc- tion of microsatellite-based linkage maps and identification of size-related quantitative trait loci for Zhikong scallop ()., 40: 821-831.
Zhan, X., Fan, F. L., You, W. W., Yu, J. J., and Ke, C. H., 2012. Construction of an integrated map ofusing microsatellite markers., 14: 79- 86.
Zhong, X. X., Li, Q., Guo, X., Yu, Y., and Kong, L. F., 2014. QTL mapping for glycogen content and shell pigmentation in the Pacific oysterusing microsatellites and SNPs., 22: 1877-1889.
(Edited by Qiu Yantao)
(Received May 4, 2017; revised July 11, 2017; accepted April 11, 2018)
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2018
# These authors contributed equally to this study.
E-mail: lxq6808@163.comE-mail: yingpan@gxu.edu.cn
杂志排行

Journal of Ocean University of China的其它文章
- Offshore Fault Geometrics in the Pearl River Estuary, Southeastern China: Evidence from Seismic Reflection Data
- Application of Geoid Anomalies to the Tectonic Research in the East Asian Continental Margin
- Middle Holocene Organic Carbon and Biomarker Records from the South Yellow Sea: Relationship to the East Asian Monsoon
- Mesozoic Deformation and Its Geological Significance in the Southern Margin of the South China Sea
- Optimization of Shanghai Marine Environmental Monitoring Sites in the Identification of Boundaries of Different Water Quality Grades
- Seasonal Variation of Environmental Variables and Phytoplankton Community Structure and Their Relationship in Liaodong Bay of Bohai Sea, China