Genome-Wide Scan for Positively Selected Genes in SessileMolluscs Highlights the Genetic Basis for TheirAdaptation to Attached Lifestyle
2018-08-24YUHongandLIQi
YU Hong, and LI Qi, *
Genome-Wide Scan for Positively Selected Genes in SessileMolluscs Highlights the Genetic Basis for TheirAdaptation to Attached Lifestyle
YU Hong1), 2), and LI Qi1), 2), *
1),,,266003,2),,266003,
Molluscs are one of the most diverse groups of animals and exhibit a rich and diverse variety of form and lifestyle. Most molluscs live with a free-moving lifestyle, while some molluscs are sessile. The adaptation to the two distinct lifestyles required complex changes, from molecules to organs, and physiology to morphology. In this study, we conducted a genome-wide scan for positive selection by comparing the available genomes of two sessile molluscs with four free-moving molluscs. A total of 40 genes were identified undergoing positive selection in the sessile molluscs by the branch-site model. Functional characterization showed that they were mainly enriched in two pathways, Oxidative phosphorylation (OXPHOS) and TGF-beta signaling pathway. The unexpected positive selection on OXPHOS genes in sessile molluscs suggests that the adaptation of OXPHOS involves many factors beyond enhancing ATP production. A modified OXPHOS regulatory system may allow sessile molluscs to better cope with biotic and abiotic stresses. Moreover, positively selected genes in TGF-beta signaling pathway probably have played a key role in the patterning of body plans and growth in metazoans. We speculate that these genes are associated with the body structure and organic adaptation to a sedentary lifestyle in sessile bivalve molluscs.
molluscs; adaptation evolution; positive selection; sessile lifestyle
1 Introduction
Mollusca is the second largest phylum in the animal kingdom, and one of the most diverse groups of animals. Molluscs exhibit a rich and diverse variety of form and lifestyle. The molluscan body plan is enormously varied, and extremely plastic and adaptable, which has led to their remarkable evolutionary success, representing about 200000 living species (Ponder and Lindberg, 2008). Based on the moving ability, molluscs can be categorized into free-moving and sessile forms. In general, most of the molluscs are free-moving (., squids, cuttlefishes, snails, and sea slugs), while some molluscs, such as adult oysters and mussels, cannot move or only move a little, and are cemented or byssally attached. Different types of lifestyles call for different physiological and anatomical adaptations such as body shape and energy metabolism (Shen., 2010; Shen., 2012). The adaptations can occur from molecules to organs. In molluscs, most sessile species are filter feeding with hypertrophied gills (Ponder and Lindberg, 2008), and their head and foot have degen-erated. Changes in energy metabolism have inevitably occurred between the sessile and free-moving molluscs. The overall rates of energy expenditure by the sessile filter-feeding species are lower than those by the browsing species (Bayne and Newell, 1983). Sedentary molluscs cannot escape predation, so they usually protect themselves using thick calcareous shells (Zhang., 2012a). In addition, some sessile filter-feeding molluscs, such as oysters inhabiting in estuaries and intertidal zones, must cope with harsh and dynamically changing environments, including wide fluctuations of temperatures and salinities as well as prolonged air exposure, and may also have to face tremendous exposure to microbial pathogens (Zhang., 2012a; Guo., 2015). Their success of adapting to different stresses suggests that sessile molluscs may possess special mechanisms for host- defense against biotic and abiotic stresses (Guo., 2015). Therefore, the sessile molluscs offer an interesting model for studying the adaptation evolution.
Many genome-wide studies have identified the selective imprints of the development of new lifestyles or locomotive styles adapting to their new ecological niches, such as lipid transport in the aquatic adaptation of cetaceans, flight adaptation in bats, transformation of nocturnal to diurnal circadian rhythm in mammals, seasonal and non-seasonal reproductive behaviors (Shen., 2012; Sun., 2013; Nery., 2013; Cao., 2015; Meng., 2015). However, our current understanding of selective imprints is mainly limited to mammals. The molluscs are one of the most diverse phyla on Earth and play an important role as model organisms in scientific research (Ponder and Lindberg, 2008), but their genetic adaptation to ecological niches is largely unknown. Re- cently, emerging high-throughput sequencing technology and bioinformatics toolshave undoubtedly facilitated the study on the genetic basis of how molluscs adapt to di- verse lifestyles. To date, six molluscan genomes (the Pa- cific oyster, pearl oyster, freshwater snail, owl limpet, California two-spot octopus, and California sea hare) have been released (Zhang., 2012a;Takeuchi, 2012; Simakov., 2013; Albertin., 2015). These six molluscs, including four free-moving and two sessile species, can represent the end-lines of different and di- verse evolutionary paths. The Pacific oyster and pearl oyster are representative bivalve molluscs. They are ses- sile filter-feeders with a bivalved shell.,andbelong to the Gastropoda, which use the foot for crawling (Heller, 2015). Octopuses, a major group of the molluscan class Cephalopoda, have changed dramatically over the course of their evolution. They are active, resourceful predators with a rich behav- ioral repertoire (Albertin, 2015). The availability of molluscan genomes provides a unique opportunity to elucidate the adaptive evolution of the sessile molluscs at the genome level.
Here, we conducted genome-wide scans for the available molluscan genome sequences to identify the genes showing evidence of adaptive evolution along the sessile mollusc lineage. The findings will help elucidate the un- derlying molecular basis of adaptation to the sessile lifestyle.
2 Methods
The protein-coding sequences for five molluscan spe- cies (,,,, and) were retrieved from the genome database in GenBank. The sequences forwere downloaded from MarinegenomicsDB (http://marinege- nomics.oist.jp/pearl/viewer/info?project_id=36).
To identify 1:1:1:1:1:1 gene orthologs, reciprocal BLASTP searches were performed on the six molluscan protein sequences (-value≤1e−5). If a gene had multiple transcripts, the longest one was chosen for further analysis. Totally 303 one-to-one orthologous genes were identified in the genomes of all the six species. The program ParaAT (Zhang., 2012b) was used to codon-align all of the 303 gene sets. To reduce the rate of false positive prediction, all gaps and ‘N’ were deleted from the alignments. In addition, the degree of nucleotide substitution saturation for each gene was tested using the method developed by Xia(2003) that has been implemented in DAMBE software (Xia, 2013). Only genes with significantly lowerthanwere retained. If the remaining alignment was shorter than 150bp (50 codons), the entire alignment was discarded. Our final data set included 286 genes. The phylogenetic tree was constructed based on the 286 genes using the maximum likelihood method in PHYML 3.0 (Guindon., 2010). The best-fit model of amino acid substitutions was selected using ProtTest 3 (Darriba., 2011)and Akaike Information Criterion (AIC). The substitution model LG+I+G had the smallest AIC value and was therefore the most appropriate substitution model. The resulting phylogenetic tree (Fig.1) had the same branching order as the tree developed by Kocot(2011). The 286 aligned protein-coding sequences and the phylogenetic tree estimated by PHYML 3.0 were used to estimate the parameters and test the hypotheses about the adaptive evolution in PAML’s codeml program (Yang, 2007).

Fig.1 Phylogenetic tree of six molluscan species used for the screen for PSGs. The blue line represents the sessile molluscs lineage. Branch of interest (sessile molluscs) is marked as S.
We used one-ratio model in PAML to estimate the distribution of the nonsynonymous/synonymous substitution ratios (=d/d) as a benchmark under an assumption of no adaptive evolution in the gene sequences and free-ratio model to estimate parameters for the genes of the labeled branch of the tree. The improved branch-site model was applied to identify genes under positive selection on a specific branch of the tree (Zhang, 2005). In this case, the lineage leading to the two sessile bivalves was labeled as foreground branch (branch S, Fig.1) and the others as background branches. A likelihood ratio test (LRT) compared a model with positive selection on the foreground branch to a null model where no positive selection occurred on the foreground branch and the statistic (2Δln) was calculated to obtain avalue (Yang and Dos Reis, 2011). Thevalue correction was performed based on the method of Benjamini-Hochberg (Benjamini and Hochberg, 1995). In our study, genes whose LRT χ2analysis yielded-values <0.05 and FDR<0.2 were considered to be positively selective gene (PSG) candidates. To gain insight in the putative functions of the PSGs found on the lineage S, theortholog of each PSG was used to perform Gene Ontology terminology (GO) and KEGG pathway annotation for all the PSGs using the web-based OmicShare tools (www.omicshare.com/tools). GO and KEGG enrichment analyses were estimated by hypergeometric test, and the calculated-value was gone through FDR correction, taking FDR<0.05 as a threshold.
3 Results and Discussion
Totally 303 single copy 1:1 orthologs from six available molluscan genomes were identified. Saturation of nucleotide substitution can bias evolutionary inferences, and result in an underestimation of dand an inflation of d/d(). Therefore, we only retained 286 orthologs without significant substitution saturation for further analysis. In order to estimate the impact of natural selection acting on genes, the one-rate model, which provides a single estimate ofacross all codons and lineages, was utilized. All the 286 orthologs hadvalues in the range of 0.001–0.15, and no extremevalue was detected. The branch model (model=2) was used to obtain the d/dvalues for all the genes on the labeled branch (branch S, Fig.1). The results showed that 61.19% of the genes hadvalues larger than 1 (Fig.2), suggesting a dramatic change in the selective pressures acting on the genes on branch S, thereby providing potential explanations for the emergence of new body plans, organs and lifestyles on branch S.
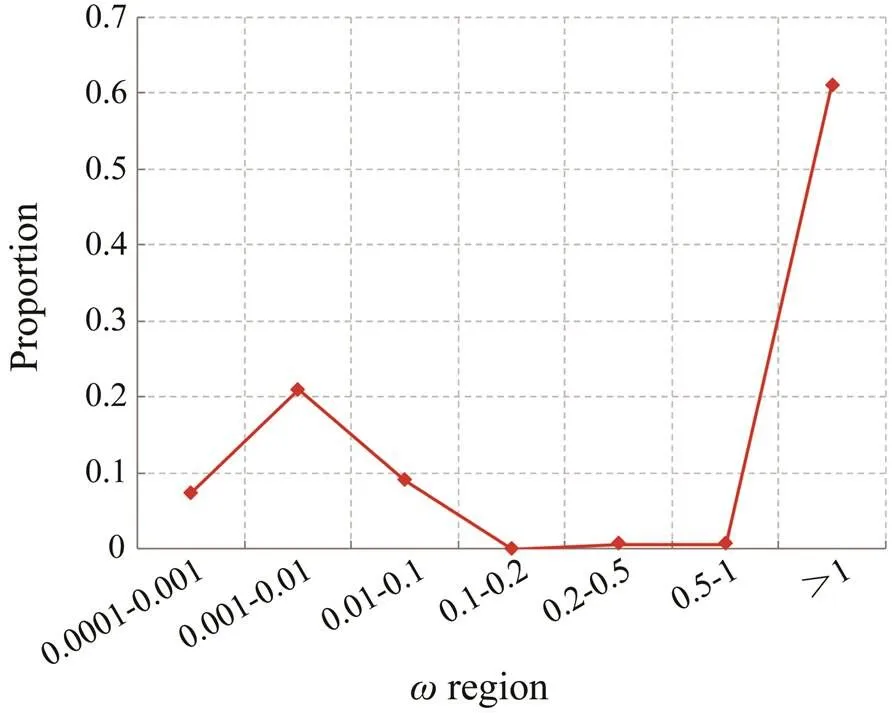
Fig.2 Distribution of ω values obtained for all the genes on branch S using the branch model. Proportion of genes in the different ω regions are indicated by the Y-axis with the ω regions on the X-axis.
To detect signals of positive selection that may have occurred on branch S, the improved branch-site model was used in this study. This model can detect positive selection acting on a few sites on particular specified lineages. As a result, 40 genes showed significant evidence of positive selection (FDR<0.2) on branch S. In order to understand the biological significance of the PSGs, we classified them according to the scheme established by the Gene Ontology project. Of the 40 PSGs, 22 PSGs were classified into 17 GO terms (Fig.3). The result revealed the most GO hits were in ‘cellular component’, followed by ‘biological process’ and ‘molecular function’. Within the ‘biological process’ category, the most GO hits were in ‘metabolic process’. Nine PSGs were related to catalytic activities within the molecular function category, including transferase activity, oxidoreductase activity, and hydrolase activity.

Fig.3 Histogram of the GO annotations of PSGs. The X-axis represents GO terms belonging to three categories and the Y-axis represents gene numbers of each term.
Among the 40 PSGs, 13 PSGs were assigned to ten pathways (Table 1). Enrichment analysis revealed that two pathways, Oxidative phosphorylation (OXPHOS) and TGF-beta signaling pathway, were significantly enriched among the PSGs. Surprisingly, genes NDUFB10, ATPeF0B and Cyt1 were involved in Oxidative phosphorylation pathway under positive selection inand. This result was in strong contrast to most previous studies which detected association ofpositive selection on OXPHOS genes with evolution of a variety of energetically demanding characteristics (Shen., 2010; Shen., 2012). A common expectation is that OXPHOS functional efficiency is important to energy intensive processes, and positive selection on OXPHOS genes would lead to enhanced organismal performance, such as powered flight (Shen., 2010). However, strong positive selection on OXPHOS genes detected among fishes with low aerobic performance did not match simple expectations, suggesting that the adaptation of OXPHOS involved many factors beyond enhancing ATP production in high performance taxa, and would not be readily predicted based solely on organismal performance (Zhang and Broughton, 2015). In this study, the result showing positive selection on OXPHOS genes in sessile molluscs was consistent with those in low-performance fishes. The organismal fitness in sessile molluscs may be increased by a modified OXPHOS regulatory system or even a reduction in OXPHOS efficiency.

Table 1 Results of pathway enrichment analysis of PSGs from branch S
TGF-beta signalingplays a key role in the patterning of metazoan body plans and growth(Kenny., 2014). It is widely regarded as a ‘module’ capable of co-option into novel functions. In gastropods, TGF-beta signaling pathway has been reported as a major driving force of torsion by regulating asymmetric cell proliferation (Kurita and Wada, 2011). During the evolution of molluscs leading to bivalves, the most prominent morphological change was that a single dorsal shell was doubled (Kin, 2009). A member of TGF-beta family,, was found to be very important for establishing the characteristic shape of the shell anlagen in Japanese spiny oyster, suggesting that TGF-beta signaling pathway played a pivotal role in the morphological transition to two shells in bivalve (Kin., 2009). In this study, two genes (BAMBI and ID1) in TGF-beta signaling pathway were found undergoing po- sitive selection, which may be associated with the great changes in body plans and organs of the sessile molluscs.
A little unexpectedly, we did not find any known genes involved in immune and stress responses under positive selection in the two sessile bivalves, considering they may face tremendous exposure to microbial pathogens and inhabit in highly stressful and widely changing environments. This result is probably because only single copy 1:1 orthologs were selected to identify the natural selection in our study. Recent studies have revealed massive expansion of immune genes in sessile bivalves, suggesting that the expansion was also important for the evolution and adaptation of bivalve molluscs (Zhang., 2012a; Guo., 2015; Takeuchi., 2016).
4 Conclusions
In summary, we conducted the first genome-wide scan for positively selected genes to investigate the genetic basis of attached lifestyle in the sessile molluscs, providing a new insight into the molecular landscape of sessile adaptation in molluscs. Although only six molluscan species were included in the analysis, it is clear that positive selection is indeed an important source of evolutionary innovation and plays a critical role in the evolution of the sessile molluscs. Along with the development of the genome sequencing techniques, assembly of higher-quality genomes, and more accurate gene annotation, the availability of more molluscan genomes will provide more resources for the investigation of adaption evolution in molluscs.
Acknowledgements
This study was supported by the grants from the National Natural Science Foundation of China (No. 3137 2524), the Shandong Seed Project, Shandong Province (No. 2014GHY115002), the Fundamental Research Funds for the Central Universities (No. 201762014), and Qingdao National Laboratory for Marine Science and Technology (No. 2016LMFS-A06).
Albertin, C. B., Simakov, O., Mitros, T., Wang, Z. Y., Pungor, J. R., Edsinger-Gonzalez, E., Brenner, S., Ragsdale, C. W., and Rokhsar, D. S., 2015. The octopus genome and the evolution of cephalopod neural and morphological novelties., 524: 220-224.
Bayne, B. L., and Newell, R. C., 1983. Physiological energetics of marine molluscs. In:4. Saleuddin, A. S. M., and Wilbur, K. M., eds., Academic Press, New York, 407- 515.
Benjamini, Y., and Hochberg, Y., 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing.–, 57: 289-300.
Cao, X., Sun, Y. B., Irwin, D. M., Wang, G. D., and Zhang, Y. P., 2015.Nocturnal to diurnal transition in the common ancestor of haplorrhines: Evidence from genomic-scan for positively selected genes., 42: 33-37.
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D., 2011. ProtTest 3: Fast selection of best-fit models of protein evolution., 27: 1164-1165.
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O., 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0.,59: 307-321.
Guo, X., He, Y., Zhang, L., Lelong, C., and Jouaux, A., 2015. Immune and stress responses in oysters with insights on adaptation., 46: 107-119.
Heller, J., 2015.. Springer, New York, 354pp.
Kenny, N. J., Namigai, E. K. O., Dearden, P. K., Hui, J. H. L., Grande, C., and Shimeld, S. M., 2014. The Lophotrochozoan TGF-β signalling cassette-diversification and conservation in a key signalling pathway.,58: 533-549.
Kin, K., Kakoi, S., and Wada, H., 2009. A novel role forin the shaping of bivalve shells revealed in a conserved molluscan developmental program., 329: 152- 166.
Kocot, K. M., Cannon, J. T., Todt, C., Citarella, M. R., Kohn, A. B., Meyer, A., Santos, S. R., Schander, C., Moroz, L. L., Lieb, B., and Halanych, K. M., 2011. Phylogenomics reveals deep molluscan relationships., 477: 452-456.
Kurita, Y., and Wada, H., 2011. Evidence that gastropod torsion is driven by asymmetric cell proliferation activated by TGF-β signaling.,7: 759-762.
Meng, Y., Zhang, W., Zhou, J., Liu, M., Chen, J., Tian, S., Zhuo, M., Zhang, Y., Zhong, Y., Du, H., and Wang, X., 2015. Genome-wide analysis of positively selected genes in seasonal and non-seasonal breeding species.,10 (5): e012 6736.
Nery, M. F., González, D. J., and Opazo, J. C., 2013. How to make a dolphin: Molecular signature of positive selection in cetacean genome., 8 (6): e65491.
Ponder, W. F., and Lindberg, D. R., 2008.. University of California Press, Berkeley and Los Angeles, 469pp.
Shen, Y. Y., Liang, L., Zhu, Z. H., Zhou, W. P., Irwin, D. M., and Zhang, Y. P.,2010. Adaptive evolution of energy metabolism genes and the origin of flight in bats.,107: 8666-8671.
Shen, Y. Y., Zhou, W. P., Zhou, T. C., Zeng, Y. N., Li, G. M, Irwin, D. M., and Zhang, Y. P., 2012. Genome-wide scan for bats and dolphin to detect their genetic basis for new locomotive styles., 7 (11): e46455.
Simakov, O., Marletaz, F., Cho, S. J., Edsinger-Gonzales, E., Havlak, P., Hellsten, U., Kuo, D. H., Larsson, T., Lv, J., Arendt, D., Savage, R., Osoegawa, K., de Jong, P., Grimwood, J., Chapman, J. A., Shapiro, H., Aerts, A., Otillar, R. P., Terry, A. Y., Boore, J. L., Grigoriev, I. V., Lindberg, D. R., Seaver, E. C., Weisblat, D. A., Putnam, N. H., and Rokhsar, D. S.,2013. Insights into bilaterian evolution from three spiralian genomes., 493: 526-531.
Sun, Y. B., Zhou, W. P., Liu, H. Q., Irwin, D. M., Shen, Y. Y., and Zhang, Y. P., 2013. Genome-wide scans for candidate genes involved in the aquatic adaptation of dolphins.,5: 130-139.
Takeuchi, T., Kawashima, T., Koyanagi, R., Gyoja, F., Tanaka, M., Ikuta, T., Shoguchi, E., Fujiwara, M., Shinzato, C., Hisata,
K., Fujie, M., Usami, T., Nagai, K., Maeyama, K., Okamoto, K., Aoki, H., Ishikawa, H., Masaoka, T., Fujiwara, A., Endo, K., Endo, H., Nagasawa, H., Kinoshita, S., Asakawa, S., Watabe, S., and Satoh, N., 2012. Draft genome of the pearl oyster: A platform for understanding bivalve biology.,19 (2): 117-130.
Takeuchi, T., Koyanagi, R., Gyoja, F., Kanda, M., Hisata, K., Fujie, M., Goto, H., Yamasaki, S., Nagai, K., Morino, Y., Miyamoto, H., Endo, K., Endo, H., Nagasawa, H., Kinoshita, S., Asakawa, S., Watabe, S., Satoh, N., and Kawashima, T., 2016. Bivalve-specific gene expansion in the pearl oyster genome: implications of adaptation to a sessile lifestyle., 2: 3.
Xia, X., 2013. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution.,30: 1720-1728.
Xia, X., Xie, Z., Salemi, M., Chen, L., and Wang, Y., 2003. An index of substitution saturation and its application.,26: 1-7.
Yang, Z., 2007. PAML 4: Phylogenetic analysis by maximum likelihood., 24: 1586-1591.
Yang, Z., and Dos Reis, M., 2011. Statistical properties of the branch-site test of positive selection., 28: 1217-1228.
Zhang, F., and Broughton, R. E., 2015. Heterogeneous natural selection on oxidative phosphorylation genes among fishes with extreme high and low aerobic performance., 15: 173.
Zhang, G., Fang, X., Guo, X., Li, L., Luo, R., Xu, F., Yang, P., Zhang, L., Wang, X., Qi, H., Xiong, Z., Que, H., Xie, Y., Holland, P. W. H., Paps, J., Zhu, Y., Wu, F., Chen, Y., Wang, J., Peng, C., Meng, J., Yang, L., Liu, J., Wen, B., Zhang, N., Huang, Z., Zhu, Q., Feng, Y., Mount, A., Hedgecock, D., Xu, Z., Liu, Y., Domazet-Lošo, T., Du, Y., Sun, X., Zhang, S., Liu, B., Cheng, P., Jiang, X., Li, J., Fan, D., Wang, W., Fu, W., Wang, T., Wang, B., Zhang, J., Peng, Z., Li, Y., Li, N., Wang, J., Chen, M., He, Y., Tan, F., Song, X., Zheng, Q., Huang, R., Yang, H., Du, X., Chen, L., Yang, M., Gaffney, P. M., Wang, S., Luo, L., She, Z., Ming, Y., Huang, W., Zhang, S., Huang, B., Zhang, Y., Qu, T., Ni, P., Miao, G., Wang, J., Wang, Q., Steinberg, C. E. W., Wang, H., Li, N., Qian, L., Zhang, G., Li, Y., Yang, H., Liu, X., Wang, J., Yin, Y., and Wang, J.,2012a. The oyster genome reveals stress adaptation and complexity of shell formation., 490: 49-54.
Zhang, J., Nielsen, R., and Yang, Z., 2005. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level., 22: 2472-2479.
Zhang, Z., Xiao, J., Wu, J., Zhang, H., Liu, G., Wang, X., and Dai, L.,2012b.ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments., 419: 779-781.
(Edited by Qiu Yantao)
(Received August 3, 2017; revised November 13, 2017; accepted April 10, 2018)
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2018
. Tel: 0086-532-82031622E-mail: qili66@ouc.edu.cn
杂志排行

Journal of Ocean University of China的其它文章
- Offshore Fault Geometrics in the Pearl River Estuary, Southeastern China: Evidence from Seismic Reflection Data
- Application of Geoid Anomalies to the Tectonic Research in the East Asian Continental Margin
- Middle Holocene Organic Carbon and Biomarker Records from the South Yellow Sea: Relationship to the East Asian Monsoon
- Mesozoic Deformation and Its Geological Significance in the Southern Margin of the South China Sea
- Optimization of Shanghai Marine Environmental Monitoring Sites in the Identification of Boundaries of Different Water Quality Grades
- Seasonal Variation of Environmental Variables and Phytoplankton Community Structure and Their Relationship in Liaodong Bay of Bohai Sea, China