A Population Genetic Analysis of Continuously SelectedChlamys farreri Populations
2018-08-24ZHANGLuLIYuliLIYangpingYANGZhihuiLIYuqiangWANGYangfanWANGShiandBAOZhenmin
ZHANG Lu, LI Yuli, LI Yangping, YANG Zhihui, LI Yuqiang, WANG Yangfan, WANG Shi, and BAO Zhenmin
A Population Genetic Analysis of Continuously SelectedPopulations
ZHANG Lu, LI Yuli*, LI Yangping, YANG Zhihui, LI Yuqiang, WANG Yangfan, WANG Shi, and BAO Zhenmin
,,,266003,
This study applied an optimized one-step 2b-RAD library construction strategy and performed simplified genome sequencing of 539 individuals from three continuously selectedpopulations. SNP screening was performed using RAD typing software and population genetic parameters for the continuously selected populations from three generations (G1, G2, G3) were determined. The results showed that the optimized one-step 2b-RAD library construction strategy greatly simplified the experimental process, making it suitable for efficiently constructing a large number of libraries. A total of 18450 SNP markers were identified, which evenly distributed throughout the genome. Population genetic analysis of these three generations showed that the mean value of observed heterozygosity was 0.275±0.177, 0.272±0.181 and 0.275±0.166, respectively. Meanwhile, the mean value of expected heterozygosity was 0.275±0.141, 0.274±0.145 and 0.280±0.133, respectively. The Wright’s fixation index (F) was 0.04291, 0.04976 and 0.06685, respectively. Markers deviated from Hardy–Weinberg equilibrium accounted for 10.34%, 12.64%, and 23.11%, and the Shannon diversity index was 0.0999±0.0404, 0.0921±0.0388 and 0.0733±0.0308.IS(also known as the inbreeding coefficient) of the three populations was 0.0256, 0.0323 and 0.0468, respectively. We suggested that the 2b-RAD method is well suited to population genetic studies of aquacultured organisms. Moreover, our results indicated that the continuous selection affected the population genetic structure of the cultured Penglai-Red scallop, but the change was not significant; therefore, population selection should continue.
2b-RAD; population genetic;; SNP
1 Introduction
Aquaculture organisms generally have a great fecundity, and only a small number of parents are needed for progeny expansion and meeting production requirement. The lack of scientific selection or scientific breeding programs may result in significantly decreased genetic diversity of the entire breeding population.The accumulating genetic loads might lead to inbreeding depression for economic traits. Therefore, during selective breeding, the high population genetic diversity should be maintained. Population genetic diversity analysis of aquaculture organisms using molecular markers can directly identify polymorphisms from the genetic materials of the population. The main method is to screen for polymorphic molecular markers and conduct population genetic diversity analysis of a species or a population. By assessing the genetic diversity and structure of a population, the degree of variation and the breeding value can be determined, ultimately guiding scientific breeding measures.
There have been many reports on using molecular mar- kers for conducting population genetic analysis of aquaculture organisms. For example, five pairs of AFLP selective primers were used to evaluate the genetic diversity of the yellow croaker populations in Qingdao and Xiamen of China, and calculated the proportion of polymorphic loci andand Shannon diversity indices (Han., 2006). The results suggested that there was no significant genetic difference between the two populations, and there existed obvious gene exchange. Eight microsatellite loci were used to analyzing the effective number of alleles and the average observed heterozygosity for fivescallop populations (Chang., 2007). A Hardy-Weinberg (H-W) equilibrium test and an F-test suggested that the five populations had different degrees of deviation from the equilibrium. Though the degree of differentiation between populations was high, most genetic variations came from individuals within the same population. Recently, in the research of the North Atlantic Ocean planktonic copepod,, restriction site-associated DNA (RAD) sequencing on a PROTON platform was used and the analysis of RAD tags showed a significant structure across the North Atlantic Ocean (Blanco-Bercial and Bucklin, 2016). Similar studies have also been performed in(Miller., 2011),(Zhan., 2009),(Li., 2016),(Bohling., 2016),(Wang., 2015),(O’Don- nell., 2014),(Liu., 2006), and(Janson., 2015).
The rapid development of high-throughput sequencing technology makes the use of SNP in genome-wide population genetic analysis possible. Compared with the traditional SNP development methods which are high-cost and inefficient, high-throughput sequencing techniques can identify a large number of SNP loci at a low cost. Various methodological approaches exist, but reduced-represen- tation genome sequencing is particularly suitable for aquaculture organisms that do not have a reference genome. Restriction sites associated DNA sequencing (RAD) and genotyping by sequencing (GBS) are common techniques for genome simplification (Davey., 2010). Recently,2b-RAD, an easier method was developed, with simplified techniques and experimental processes (Wang., 2012). Meanwhile, with the basis of 2b-RAD me- thod, maximum likelihood (iML) algorithm (Dou., 2012) and RAD typing software package (Fu., 2013) were developed, which increased the genotyping accuracy and could be employed in identifyinggenotype markers. The operation is flexible and simple, and is suitable for the large-scale development and application of SNP mar- kers for non-model organisms. For example, in the bivalve mollusk, a high-resolution genetic linkage map was established to identify QTLs for sex determination and several economic traits (Jiao., 2014). In Chinese mitten crabthe high density linkage map was established and the high-density genetic mapping results indicated a ZW sex determining system (Cui., 2015)
As an important mariculture shellfish in China,is native to the coastal areas of China, Japan, and South Korea. In the 1970s, with the introduction of semi- artificial collection and artificial breeding techniques, large-scale farming ofwas developed in China. With the expansion of aquaculture scale, elite strains with high yield and high stress-resistance are urgently desired in response to the challenges brought by blind propagation and the decreased quality of aquaculture environment. A number of studies onhave been carried out. High-yield and high-resistance strains ofandhave been developed. With the development of high-throughput sequencing technology and reduced technical costs, the application of a large number of SNP markers in the population genetic analysis ofcan be realized, which will provide the theoretical basis for the research on the breeding industry of.
This study optimized the one-step 2b-RAD library construction strategy. After restriction enzyme digestion and adaptor ligation, only a single round of PCR was performed to establish the sequencing library, which simplified the experimental process, shortened the experimental time, and identified a large number of SNP loci. The obtained SNPs were used in the population genetic analysis of three continuously selected populations of. The number of loci conforming to H-W equilibrium, observed heterozygosity, expected heterozygosity, Wright’s fixation index, the Shannon diversity index, and the inbreeding coefficient (IS) were estimated.
2 Materials and Methods
2.1 Scallop Sample Collection and DNA Extraction
scallops used in this study were collected from Shazikou Bay Area, Qingdao. They belong to the breeding population of the new national strain Penglai- Red. In the years of 2011, 2013, 2015, shell height was used as a selective trait, and the top 5% of the scallops were selected. Individuals contributing gametes in all three generations were dissected to collect adductor muscles or gills, which were fixed in ethanol for subsequent experiments. Individuals were randomly selected from the three populations for genetics analysis, of them 76 from G1; 107 from G2; and 356 from G3.
Genomic DNA from a total of 539scallops of three generations was extracted using the traditional phenol/chloroform extraction method (Sambrook., 1989). DNA integrity was measured using 1% agarose gel electrophoresis, and DNA concentration was determined using Nano-View. Samples with concentration greater than 200ngµL−1were selected for subsequent library construction. To ensure the uniformity of the constructed 2b-RAD libraries, DNA samples were diluted to 100ngµL−1prior to restriction enzyme digestion.
2.2 2b-RAD Library Construction
One-step library construction strategy was optimized based on the 2b-RAD method (Wang., 2012). After restriction enzyme digestion and adaptor ligation, two pairs of PCR primers at the corresponding ratio were added to the same reaction mixture for PCR amplification. Polyacrylamide gel electrophoresis was used to recover the corresponding target fragments for high-throughput sequencing.
The experiment methods based on 2b-RAD methods (Wang., 2012). The concentration of two pairs of PCR primers were optimized. Two pairs of PCR primers were included in the PCR reaction (Slx-1ST-MpPrimer-1 and Slx-1ST-MpPrimer-2; Slx-2ND-MpPrimer and Slx- barcode). The reaction volume was 20µL, including 0.6µL 10mmolL−1(each) dNTP, 1µL 1µmolL−1Slx-1st- MpPrimer-1, 1µL 1µmolL−1Slx-1st-MpPrimer-2, 4µL 1µmolL−1Slx-2ND-MpPrimer, 4µL 1µmolL−1Slx-bar- code, 4µL 5× HF Buffer, 0.2µL Phusion polymerase, 4µL ligated DNA, and 1.2µL sterile deionized water. The PCR amplification program followed 16 cycles of 98℃ denaturation for 5s, 60℃ annealing for 20s, and 72℃ extension for 10s, followed by 72℃ extension for 10min and subsequent storage at 4℃. After the completion of the PCR reaction, 3.5µL samples from each library were analyzed using 8% polyacrylamide gel at 300V to observe whether target bands were clear.
After target bands were identified using 8% polyacrylamide gel electrophoresis, samples were quantified and mixed in equal proportions. The mixture was further subjected to 8% polyacrylamide gel electrophoresis, and target gel fragments were cut and stored in a 1.5mL EP tube. After a grinding with a pestle, 30–50µL triple distilled water was added, and the combined sample was incubated at 4℃ for 12h. These were then transferred using a pipette into a separation column and placed in a new centrifugation tube. Centrifugation was carried out at 4℃ at 12000rmin−1for 5min to recover the liquid, which constituted the pooled library.
Quality control and concentration determination for each library were performed using Qubit. Sequencing was performed by BerryGenomics, Inc. (Beijing, China). Libraries were constructed for 539 individuals and were evenly distributed in nine lanes for sequencing.

Table 1 Adaptor and primer information
2.3 Processing of the Returned Data
The sequencing was performed by BerryGenomics, Inc. using the Illumina Hiseq2000 sequencing system. The obtained sequencing data were analyzed using the RADtyping software package (Fu., 2013). The main process can be summarized as follows: 1) The adaptor sequences of all tags were removed to obtain the original raw data; 2) The BSAXI restriction enzyme recognition site with sequence N9ACN5CTCCN7 was extracted; reads without this site, containing two or more ambiguous base calls (N), containing more than 10 consecutive identical bases, or containing more than 5 low quality bases (quality<10) were considered poor data and were removed; 3) based on the obtainedgenome sequence, all thegenerated BSAXI tags were used as reference sequences, and the returned sequencing data were mapped using SOAP software; tags that had one alignment (soap data) were subjected to subsequent classification analysis.
SNP genotyping was also performed using a RADtyping software. Tags generated bydigestion ofgenome were pooled as reference sequences, and sequencing data from all individuals were mapped against the reference sequences. Parameters affecting SNP classification included -m (minor allele frequency), -g (percentage of minimum participants), -s (maximum number of SNPs per read), and they were set as -m 0.01, -g 0.8, and -s 3.
2.4 Population Genetic Parameter Estimation
2.4.1 Heterozygosity
Heterozygosity refers to the ratio of heterozygous individuals in a population, which is used for assessing genetic diversity within a population (Bao., 2011). The average expected heterozygosity is also called Nei’s genetic diversity. The formula is as follows:


2.4.2 Shannon diversity index
Shannon diversity index takes into account the number of alleles and abundance to measure the population genetic diversity (Shannon and Weaver, 1951). The formula is as follows:


2.4.3 Wright’s fixation index
Wright’s fixation index measures the degree of deviation of observed heterozygosity from the expected heterozygosity of the population, which was developed by Wright (1965):

2.4.4 Inbreeding coefficient
The inbreeding coefficientISwas calculated using PLINK software. (http://zzz.bwh.harvard.edu/plink/).
3 Experimental Results
3.1 One-Step 2b-RAD Library Construction Strategy
DNA quality directly relates to the quality of the constructed library. Therefore, DNA from each individual was subjected to agarose gel electrophoresis analysis, and DNA showing clear main bands without smearing was considered to be of sufficient quality. Diffused bands were observed for the experimental groups, while intact DNA was observed for the control groups (Fig.1).

Fig.1 Digestion with BSAXI enzyme (E) compared with control (C).
During the one-step 2b-RAD library construction, two primer pairs were added to the PCR reaction, and target fragments were obtained through one round of PCR. During the reaction, the two primer pairs amplified each other and generated non-specific bands. Previous experiments testing the results of different combinations of the two primer pairs have determined the optimal ratios, which enhanced the target fragments amplification and decreased the amplification of the non-target fragments. In this study, the target fragment size was 156bp (Fig.2). After calculating the target fragment concentration for each library, every 20 libraries were mixed in equal proportions and recovered after polyacrylamide gel electrophoresis (Fig.3).

Fig.2 PCR products of target fragment by polyacrylamide gel electrophoresis.
Fig.3 Mixed library recovered after polyacrylamide gel electrophoresis.
3.2 SNP Loci Identification
3.2.1 Data processing
In this study, libraries for 539 individuals from three generations were constructed. An average of 60 individuals were sequenced in each lane, and after sequencing completion, barcodes were used for identifying each individual. The returned data showed that an average of 57209 tags were obtained per individual with an average depth of 20×. A total of 1647845439 clean reads with 59322435804 clean bases were obtained. Of the 539 individuals, the data for three individuals, which might be due to failed library construction, were removed, resulting in sequencing information for 536 individuals. The RAD typing software package was used for SNP identification and genotyping. The results showed that among the clean reads, processing data accounted for an average of 87.6% of the total. After the mapping was performed against the constructed reference sequences, an average of 650820714 reads from an individual could be mapped to a single reference sequence, accounting for 44.83% of the processing data. Other tags were not aligned to a unique position, suggesting that thegenome contains a large number of repeated sequences. Of the 536individuals, the minimum number of tags was 44835, and the maximum number was 85050, with an average of 57209 and an average depth of 20×. Using the set parameters (-m 0.01, -g 0.8, -s 3), 18450 SNP loci were obtained, and each was with MAF>0.01 and could be classified in 80% of the individuals. The number of SNPs on each read was less than three.
3.2.2 SNP locus information
The frequency distribution of MAF of the selected populations of the threegenerations is shown in Fig.4. SNP loci in this study were filtered with MAF=0.01 during classification, and therefore the allele frequencies for all SNP loci were greater than 0.01. Loci with allele frequencies between 0.01 and 0.1 accounted for 63.19%, 64.03%, and 63.61% for the three generations, respectively. The numbers of low-frequency alleles (less than 0.1) were significantly different among the three generations (P < 0.05), and the numbers of loci with allele frequencies greater than 0.10 were not significantly different among the three generations.
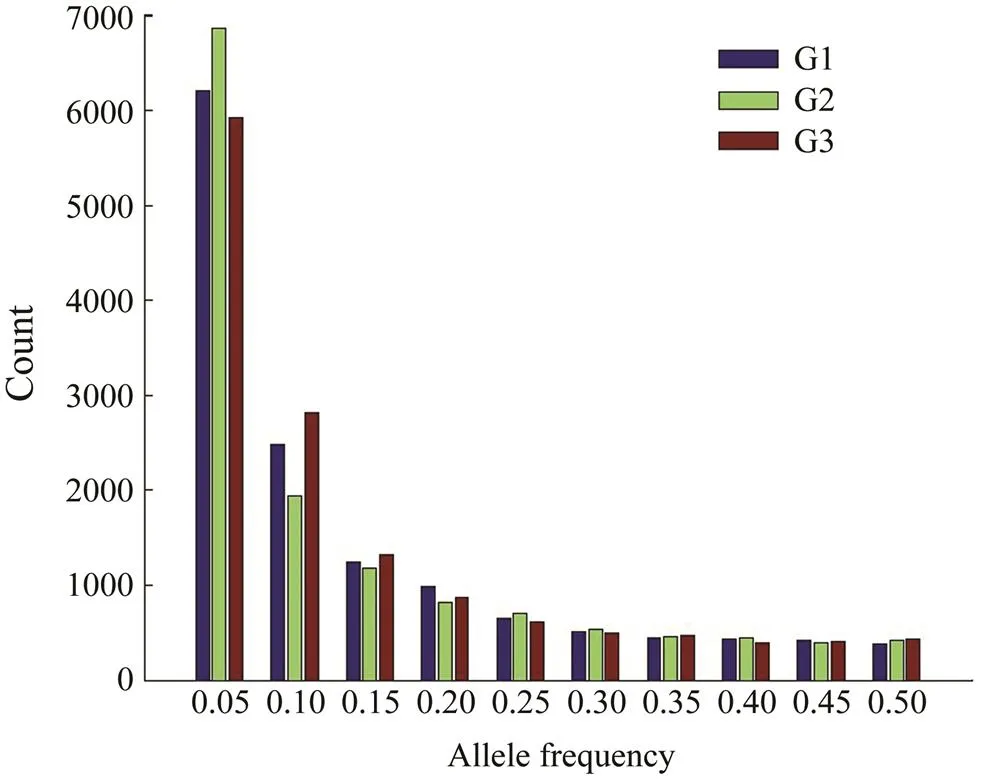
Fig.4 Distribution of minor allele frequency in three continuously selected populations.
3.3 Estimation of Population Genetic Parameters
The mean observed heterozygosity value for the continuously selected populations of the three generations was 0.275±0.177, 0.272±0.181, and 0.275±0.166, respectively; and the expected heterozygosity value was 0.275±0.141, 0.274±0.145, and 0.280±0.133, respectively.
The calculated Wright’s fixation index (F) was 0.04291, 0.04976, and 0.06685 for the three generations, respectively. A total of 18450 loci from the selected populations of the three generations were tested for H-W equilibrium, and the results showed the numbers of loci that did not conform to H-W equilibrium were, respectively, 1908, 2333, and 4263 (<0.05), accounting for 10.34%, 12.64%, and 23.11% of the total, showing an increasing trend. The distribution ofvalue for H-W equilibrium for the three generations is shown in Fig.5.

Fig.5 Histogram of Hardy-Weinberg equilibrium P-value in three continuously selected generation populations.

Fig.6 Variation of Shannon index in three continuously selected generation populations.

Fig.7 Variation of FIS value in three populations.
The Shannon diversity index of the three generations was progressively reduction.ISvalue of the three generations are showing a progressively increasing trend. The observed homozygosity, expected homozygosity, andISvalues of the three generations are shown in Table 2. The trends are shown in Figs.6 and 7.

Table 2 Homozygous observation, homozygous expectation, non-missing genotype, and FIS in three populations
4 Discussion
4.1 Optimized One-Step 2b-RAD LibraryConstruction Strategy
The previous methodology for typical 2b-RAD library construction was a two-step process. After enzyme digestion, ligation, and first-round PCR amplification, tags containing the restriction enzyme recognition site were enriched and recovered, followed by a second-round PCR for fragment extension to obtain a full-length library. The present study streamlined the two-step process into a single step, which greatly simplified the workflow, especially suited for sequencing a large number of individuals. The obtained PCR products were detected using polyacrylamide gel electrophoresis, which were mixed in equal proportions, extracted from the gel, and quantified in preparation for high-throughput sequencing.
Each individual was identified by a barcode introduced by primer slx-barcode. To ensure the quality of each library, after PCR reaction, each library was individually examined by polyacrylamide gel electrophoresis. The concentrations of target fragments for each library could be determined using QuantityOne software, and the success of library construction could be directly visualized. Barcodes were introduced during PCR amplification. During the sequencing process, each individual in each lane needed a different barcode. Barcodes containing balanced base composition for all loci of all individuals were needed to avoid amplification bias.
4.2 C. farreri SNP Loci Identification
A total of 18450 SNP markers were obtained using RADtyping software. Our group has previously conducted a systemic study on the high-resolution linkage map for, with 3806 SNP markers mapped to 19 linkage groups, an average marker interval of 0.41cM, and genome coverage reaching 99.5%, which is the first high-accuracy genetic map for bivalve mollusks (Jiao., 2014). Our group recently generated a sequence assembly for thegenome and obtained 143162 scaffolds. All SNP markers in the genetic map were mapped to all scaffolds. Scaffolds were then sorted according to the genetic distances between markers on the genetic map. All the obtained scaffolds were used as a reference, and 18450 SNP markers identified in this study were mapped to the reference sequences to obtain the positional information for all the SNP markers. Finally, 13750 SNP markers could be mapped to the corresponding linkage groups. The relative physical distances between markers are shown in Table 3.

Table 3 Distribution of marker position and number of markers in each linkage group
Among the 19 linkage groups, the maximum number of SNPs was 1051, and the minimum number of SNPs was 405; the maximum marker interval was 41.69kb, and the minimum marker interval was 32.4kb, with an average of 37.7kb. These results indicate that the one-step 2b-RAD library construction strategy identified evenly distributed SNPs in the genome, essentially covering the whole genome of.
4.3 Population Genetic Parameter Estimation
Wright’s fixation index (F) gradually increased as generations progressed, suggesting an ongoing selection, and the three generations gradually deviated from H-W equilibrium. This study tested the selected populations from the three generations for H-W equilibrium, and the data verified the effects of selection on H-W equilibrium. The Shannon diversity index of the three generations showed a gradually decreasing trend, indicating selection had a corresponding effect on the population, decreasing population diversity. TheISvalue increased with each generation, suggesting that during the selection process, the degree of inbreeding gradually increased.
The trends of Wright’s fixation index, proportion of loci not conforming to the H-W equilibrium, the Shannon diversity index, andISvalue indicated that artificial selection affected the aquaculture population; however, the change was not drastic. Selection resulted in the change in population structure, but short-term selection did not have a strong impact on the overall population structure. The results suggestted that Penglai Red scallops have abundant population diversity, and elite strains can be bred through population selection without risking the possibility of inbreeding. During the breeding process, abundant phenotypic variation and genetic diversity provide options for artificial selection criteria. When population diversity remains at high levels, an appropriate increase in selection strength can accelerate genetic progression, thereby producing economic benefits more quick- ly.
5 Summary
This study applied an optimized one-step 2b-RAD library construction strategy to perform simplified genome sequencing of 539 individuals from three selected populations of. A total of 18450 SNP markers were obtained. Using these markers, population genetic parameters of the selected populations in the three generations were estimated. The results provide a strong basis for the future development of a variety of breeding strategies for elite strains ofand other aquatic organisms, which can promote healthy and sustainable development of the aquaculture elite strain industry.
Acknowledgements
We acknowledge the supports from the National Natural Science Foundation of China (Nos. 31130054 and 31472258), the AoShan Talents Program of Qingdao National Laboratory for Marine Science and Technology (No. 2015ASTP-ES02), and the Fundamental Research Funds for the Central Universities (No. 201564009).
Bao, Z. M., Wang, M. L., Li, Y., Zhang, L. L., Hu, X. L., Huang, X. T., Hu, J. J., and Wang, S., 2011. Mathematical analysis methods used in population genetics studies based on nuclear genome markers., 41 (11): 48-56 (in Chinese with English abstract).
Blanco-Bercial, L., and Bucklin, A., 2016. New view of population genetics of zooplankton: Rad-seq analysis reveals population structure of the North Atlantic planktonic copepod., 25 (7): 1566-1580.
Bohling, J., Haffray, P., and Berrebi, P., 2016. Genetic diversity and population structure of domestic brown trout () in France., 462: 1-9.
Chang, Y. Q, Chen, X. X., Ding, J., Cao, X. B., Li, R. L., and Sun, X. W., 2007. Genetic diversity in five scallop populations of the Japanese scallop ()., 27 (3): 1145-1152.
Cui, Z., Hui, M., Liu, Y., Song, C., Li, Y., Liu, L., Shi, G., Wang, S., Li, F., Zhang, X., Liu, C., Xiang, J., and Chu, K. H., 2015. High-density linkage mapping aided by transcriptomics do- cuments ZW sex determination system in the Chinese mitten crab., 115 (3): 206.
Davey, J. W., and Blaxter, M. L., 2010. RADSeq: Next-genera- tion population genetics., 9 (5-6): 416-423.
Dou, J. Z., Zhao, X. Q., Fu, X. T., Jiao, W. Q., Wang, N. N., Zhang, L. L., Hu, X. L., Wang, S., and Bao, Z. M., 2012. Reference-free SNP calling: Improved accuracy by preventing incorrect calls from repetitive genomic regions., 7 (1): 17.
Fu, X. T., Dou, J. Z., Mao, J. X., Su, H. L., Jiao, W. Q., Zhang, L. L., Hu, X. L., Huang, X. T., Wang, S., and Bao, Z. M., 2013. RADtyping: An integrated package for accurate de novo codominant and dominant RAD genotyping in mapping populations., 8 (11): e79960.
Han, Z. Q., Gao, T. X., Wang, Z. Y., Zhuang, Z. M., and Su, T. F., 2006. Analysis of genetic diversity ofby AFLP markers., 30 (5): 640- 646.
Janson, S., Wouters, J., Bonow, M., Svanberg, I., and Olsén, K. H., 2015. Population genetic structure of crucian carp () in man-made ponds and wild populations in Sweden., 23 (1): 359-368.
Jiao, W. Q., Fu, X. T., Dou, J. Z., Li, H. D., Su, H. L., Mao, J. X., Yu, Q., Zhang, L. L., Hu, X. L., Huang, X. T., Wang, Y. F., Wang, S., and Bao, Z. M., 2014. High-resolution linkage and quantitative trait locus mapping aided by genome survey sequencing: Building up an integrative genomic framework for a bivalve mollusk., 21 (1): 85-101.
Li, H. J., Zhang, J. J., Yuan, X. T., Zhang, A. G., Liu, G. Z., Shao, K. S., and Wang, L. J., 2016. Genetic diversity and differentiation of seven geographical populations of hard clam () assessed by COI and microsatellite markers., 36 (2): 499-507.
Liu, Y. G., Chen, S. L., Li, J., and Li, B. F., 2006. Genetic diversity in three Japanese flounder () popu- lations revealed by ISSR markers., 255 (1-4): 565- 572.
Miller, P. A., Fitch, A. J., Gardner, M., Hutson, K. S., and Mair, G., 2011. Genetic population structure of yellowtail kingfish () in temperate Australasian waters inferred from microsatellite markers and mitochondrial DNA., 319 (3): 328-336.
O’Donnell, T. P., Denson, M. R., and Darden, T. L., 2014. Genetic population structure of spotted seatroutalong the south-eastern U.S.A., 85 (2): 374-393.
Sambrook, J., Fritsch, E., and Maniatis, T., 1989. Molecular cloning: A laboratory manual+ cold spring harbor., 49 (2): 411.
Shannon, C. E., and Weaver, W., 1951. The mathematical theory of communication.,26 (3): 31-32.
Wang, J., Zhang, F. Y., Jiang, K. J., Ma, C. Y., Song, W., Lin, N., Jiang, Y. Z., Li, S. F., Cheng, J. H., and Ma, L. B., 2015. Genetic diversity ofbased on the mitochondrial cytochrome oxidase subunit I sequence from the East China Sea.,37 (2): 114.
Wang, S., Meyer, E., Mckay, J. K., and Matz, M. V., 2012. 2b-RAD: A simple and flexible method for genome-wide genotyping., 9 (8): 808-810.
Wright, S., 1965. The interpretation of population structure by F-statistics with special regard to systems of mating., 19 (3): 395-420.
Zhan, A. B., Hu, J. J., Hu, X. L., Zhou, Z. C., Hui, M., Wang, S., Peng, W., Wang, M. L., and Bao, Z. M., 2009. Fine-scale population genetic structure of Zhikong scallop (): Do local marine currents drive geographical differentiation?, 11 (2): 223-235.
(Edited by Qiu Yantao)
(Received May 2, 2017; revised June 28, 2017; accepted April 1, 2018)
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2018
. E-mail: lazqd2015@126.com
杂志排行

Journal of Ocean University of China的其它文章
- Offshore Fault Geometrics in the Pearl River Estuary, Southeastern China: Evidence from Seismic Reflection Data
- Application of Geoid Anomalies to the Tectonic Research in the East Asian Continental Margin
- Middle Holocene Organic Carbon and Biomarker Records from the South Yellow Sea: Relationship to the East Asian Monsoon
- Mesozoic Deformation and Its Geological Significance in the Southern Margin of the South China Sea
- Optimization of Shanghai Marine Environmental Monitoring Sites in the Identification of Boundaries of Different Water Quality Grades
- Seasonal Variation of Environmental Variables and Phytoplankton Community Structure and Their Relationship in Liaodong Bay of Bohai Sea, China