The Impact of Irrigation on Bacterial Community Compositionand Diversity in Liaohe Estuary Wetland
2018-08-24LITiantianHUHongLIZhengyanZHANGJianyeandLIDong
LI Tiantian, HU Hong, *, LI Zhengyan, *, ZHANG Jianye, and LI Dong
The Impact of Irrigation on Bacterial Community Compositionand Diversity in Liaohe Estuary Wetland
LI Tiantian1), HU Hong1), *, LI Zhengyan1), *, ZHANG Jianye2), and LI Dong3)
1) College of Environmental Science and Engineering, Ocean University of China, Qingdao 266100, China 2) Colleget of Marine Life Sciences, Ocean University of China, Qingdao 266100, China 3) Institute of Wetland Science, Panjin 124000, China
In this study, the sequencing of 16S ribosomal DNA was used to characterize the soil bacterial community composition and diversity in Liaohe estuarine wetland. Soil samples were taken from different locations in the wetland dominated by reed. Moreover, the soil quality parameters were evaluated (pH, moisture, organic matter, total nitrogen, available nitrogen, total phosphorus, available phosphorus). The results showed that the organic matter and nutrient contents were significantly higher in irrigated wetland than those in natural wetland. Major phylogenic groups of bacteria in soil samples including Proteobacteria, Acidobacteria, Gemmatimonadetes, Actinobacteria and Cyanobacteria were analyzed and we found that Proteobacteria was the most abundant in the community, and the phylum Acidobacteria was more abundant in irrigated wetland. Beta diversity analyses indicated that the soil bacterial community was mainly affected by sampling sites rather than seasons. In general, the bacterial community in natural wetland was not significantly different with that in artificial irrigated wetland. Artificial hydraulic engineering irrigated according to the water requirement rule of reed, increased the production of reeds, changed the way of wetland soil material input, but the diversity of bacterial community kept stable relatively.
soil bacterial community; Liaohe estuary wetland;16S rDNA sequencing; nutrient
1 Introduction
Wetland ecosystem locates in the transitional zone of aquatic and terrestrial ecosystem, which has important ecological functions in nutrient cycling, energy flow, climate regulation, sediment accretion, pollutant filtration and biodiversity maintaining (Ansola., 2014). Estuarine wetlands, where freshwater meets salt water and the land connects to the sea (Barbier., 2011), show distinct ecological features, with the coexistence of fresh and salt-tolerant microorganisms, and aerobic and anaerobic microbial species. These microorganisms may be sensitive response to various environmental changes (Wang., 2012; Peralta., 2014). As one of the typical coastal wetland in China, Liaohe estuarine wetland, covering an area of 3.0×105hectares, is the largest warm temperate coastal wetland in Asia with rich biodiversity. In recent years, the area of natural wetland and soil fertility decreased because of the reduced precipitation, rise sea-level, and unreasonable utilization of water resources (Zhang., 2011; Peng., 2012). In order to improve the reed production, nearly 5.33×108m2wetlandcan be artificially irrigated by water conservancy projects. Among them, the production of reed in Yangjuanzi wetland increased from 3×104ty−1to 9×104ty−1after the Shengli irrigation district was built (Su., 2012).
Microorganism plays a key role in various ecological processes such as organic materials decomposition, nutrient transformation, and nutrient bioavailability in soil ecosystem (He., 2008; Xu., 2014; Phillips., 2015). The understanding of the soil microbial community composition and function is therefore essential for evaluating soil quality, restoring soil vitality, and promoting plant growth (Wardle., 2004; Pereira., 2013). Studies on microbial community in Liaohe estuarine were insufficient comparing with other estuarine wetlands (Yu., 2012; Zhang., 2013). In the past years, some studies on distribution of microbial community were carried out with dilution-plate method and the assessment of microbial biomass (Zhao., 2008; Zhao., 2009; Li., 2013). Impact of oil pollution on microbial community was also evaluated with DGGE method (Yin., 2014). Nevertheless, there is no report to apply 16S rDNA gene sequencing to assess the distribution of microbial community in Liaohe estuary irrigated wetland.
Recent advances in high-throughput sequencing of soil DNA allows the rapid detection of microbial communities, and allows us to study soil microbial community and its relationship with other soil properties deeply (Inceoğlu., 2011). This molecular methodology has been applied in investigating microbial community in different ecosystems, including forests, grasslands, and agricultural lands (Roesch., 2007; Suleiman., 2013; López-Piñeiro., 2013). Research of soil microbial community among different ecosystem helps to expand and deepen our knowledge on ecosystem structure and function. In wetland ecosystem, much attention has been focused on freshwater and constructed wetlands (Menon., 2013; Peralta., 2013; Ligi., 2013; Philippe., 2014). However, estuarine wetlands were seldom reported concerning the variability in microbial community structure and diversity (Wang., 2010).
This study, therefore, aims to evaluate the changes of soil bacterial community composition and diversity in irrigated and natural wetland in Liaohe estuarine, and analyze the impact of environmental factors on bacterial community diversity. So as to offer some support to reed production improvement and soil fertility recovery.
2 Materials and Methods
2.1 Study Site Description
This study was conducted in Liaohe estuarine wetland (41˚14´49´´N, 121˚47´31´´E), which is located in southwest of Liaoning province and north of Bohai Sea. This wetland is a composition delta formed by Daliao River, Liao River, and Daling River, with a total area of 3.0×105hm2. Liaohe estuarine delta is the largest warm temperate coastal wetland in China. The wetland shows a coastal sub-humid warm temperate continental monsoon climate, with an average annual temperature of 10.45℃ and an average annual rain fall of 399.5mm in 2014. The evaporation is greater than precipitation, so the wetland water mainly comes from the supply of river retention and tide (Zhang., 2011). Three fields were selected as study sites to investigate the difference in soil microbial community structure and diversity with various soil qualities. The artificial irrigated wetland (AI) is located in area of water conservancy projects, with regulated artificial irrigation of Liaohe River, Raoyang River. So the reed grows tall and have high above ground biomass (Table 1). In the natural condition, the soil basement uneven, nutritional imbalance, and inequality water distribution lead to different soil environment. Thus two sampling sites were set up. Since the natural wetland middle area (NM) is the most drought, reed depends largely on natural precipitation and ground water with little human intervention. The natural wetland edge area (NE) reeds grow better than which in NM wetland due to the better water supply.

Table 1 Indices of reed growth
Notes: Means ± standard errors,*<0.05;**<0.01;***<0.001; ns – not significant. Data followed by the same letter are not significantly different according to the LSD test (<0.05).
2.2 Soil Sampling
Soil samples were collected from wetlands in panjin, Liaoning province, three sites were selected according to the different growth situation of reed. The geographical coordinates is 121˚47´51´´E, 41˚14´81´´N; 121˚47´507´´E, 41˚14´811´´N; 121˚47´504´´E, 41˚14´612´´N. At each site, three soil samples were taken at the depth of 0-10cm from the surface by random and kept in polyethylene bags. All samples were kept in a cooler with ice packs to slow bacterial activity until further processing. In the laboratory, any visible root or plant materials were manually removed prior to homogenization, and then mixed for one pool sample. The samples were then divided into two portions. The first portion was air-dried to determine the physicochemical properties, whereas the second portion was stored under −20℃ until microbial community analysis. Soil sampling was conducted twice at each site, July and September, 2014, respectively. In July, 2014, it has the average temperature of 25.3℃, and the total sunshine hours for 155.0h. While in September 2014, it has the average temperature of 18.9℃, the average rainfall of 53.5mm, and the sunshine hours for 232.6h. The The average temperature in September was slightly lower than all the year round and had good light conditions, http: //www.panjin.gov.cn/pjnew/pjkx/tzgg/content/40288bd74ece9c05014ed740ac1172cb.html.
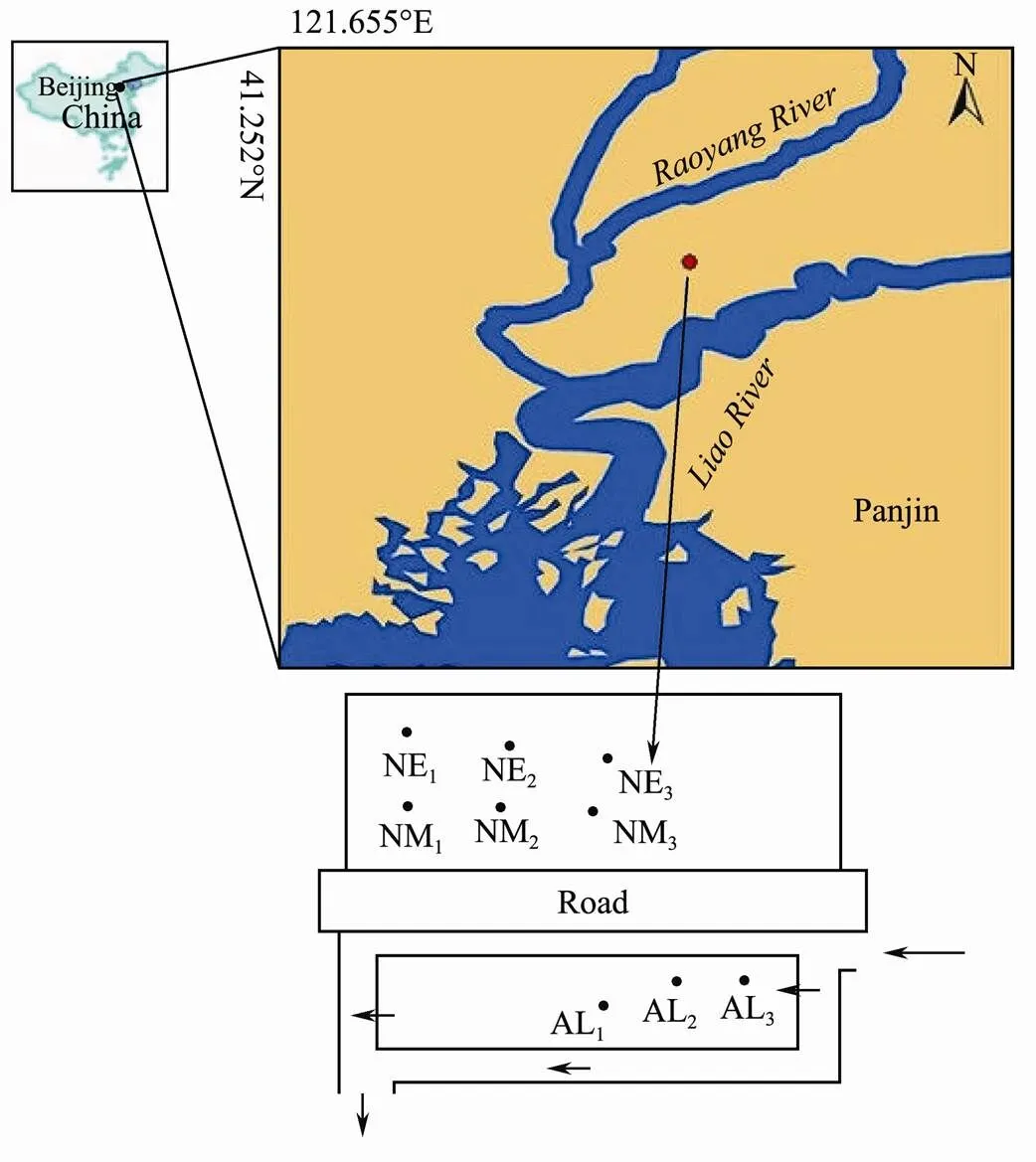
Fig.1 The map of the sampling site.
2.3 Soil Physicochemical Analysis
2.3.1 Analysis methods
All the physiochemical analysis was undertaken according to methods in soil agricultural chemical analysis (Lu, 2000). Soil moisture was measured by standard oven baking method. Soil pH was measured by pH meter with a soil water ratio of 1:2.5 (Weight:Volume). Total nitrogen (TN) and available nitrogen (N) were measured with alkaline hydrolysis diffusion method. Total phosphorus (TP) and available phosphorus (P) were measured by Mo- Sb colorimetric method. The soil organic matter (SOM) was assayed by potassium dichromate volumetry.
2.3.2 Statistics methods
Three parallel determinations were done for each sample, using Microsoft Office Excel to conduct a preliminary data processing, and SPSS19.0 to do the ANOVA of experimental data.
2.4 Microbial Analysis
Total bacterial community DNA was isolated from approximately 0.5g soil sample using the PowerSoil® DNA Isolation Kit (ANBIOSCI TECH LTD) following the manufacturer’s protocol. Products were verified by gel electrophoresis and diluted in 100μL 10m molL−1Tris. Extracted DNA was stored at﹣20℃ until further analyses.
The library was sequenced on an Illumina MiSeq platform and 250bp/ 300bp paired-end reads were generated. The 515F-806R (515F: GTGCCAGCMGCCGCGGTAA806R: GGA CTACHVGGGTWTCTAAT) primer was used to amplify the bacteria specific V4 hyper variable region of the 16S rRNA gene. All PCR reactions were carried out in 30μL reactions with 15μL of Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.2μmolL−1of forward and reverse primers, and about 10ng template DNA. Thermal cycling consisted of initial denaturation at 98℃ for 1min, followed by 30 cycles of denaturation at 98℃ for 10s, annealing at 50℃ for 30s, elongation at 72℃ for 60s, and finally 72℃ for 5min. A same volume of 1X loading buffer (contained SYB green) was mixed with PCR products and operated electrophoresis on 2% agarosegel for detection. Samples with bright main strip between 400-450bp were chosen for further experiments. PCR products were mixed in equidensity ratios. Then mixture PCR products was purified with GeneJET Gel Extraction Kit (Thermo Scientific). Sequencing libraries were generated using NEB Next® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA) following manufacturer’s recommendations and index codes were added. The library quality was assessed on the Qubit@ 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system.
Paired-end reads from the original DNA fragments were merged using FLASH, a very fast and accurate analysis tool, which was designed to merge paired-end reads when at least some of the reads overlap the reads generated from the opposite end of the same DNA fragment. Paired-end reads were assigned to each sample according to the unique barcodes. Sequences analysis was performed by UPARSE software package using the UPARSE-OTU and UPARSE-OTU ref algorithms (Edgar., 2013). In-house Perl scripts were used to analyze alpha (within samples) and beta (among samples) diversity. Sequences with ≥97% similarity were assigned to the same OTUs. We picked a representative sequence for each OTU and used the RDP classifier to annotate taxonomic information for each representative sequence (Wang., 2007). Graphical representation of the relative abundance of bacterial diversity of phylum can be visualized using Krona chart. In order to compute Alpha Diversity (Whittaker., 1972), we rarified the OTU table and calculated three metrics: Chao1 (Chao, 1987) to estimate the species abundance, observed species to estimate the amount of unique OTUs in each sample. The Shannon’s diversity index is, in whichis the proportion of individuals belonging to theth species in the data set of interest. Rarefaction curves were generated based on the three metrics. We used weighted unifrac distance for Principal Coordinate Analysis (PCoA). PCoA helps to get principal coordinates and visualize them from complex, multidimensional data. It takes a transformation from a distance matrix to a new set of orthogonal axes. The maximum variation factor is demonstrated by first principal coordinate, the second variation factor by the second principal coordinate, and so on. To identify differences of microbial communities between the two groups, ANOSIM and MRPP (multi-response permutation procedure) were performed based on the Bray-Curtis dissimilarity distance matrices (Clarke, 1993; McCune., 2002).
3 Results and Discussion
3.1 Soil Physicochemical Properties
The soil basic physicochemical properties were shown in Table 2. The pH value in the artificial irrigated wetland is 7.38, 8.08 and 8.47 in natural wetland middle and edge area, indicating that the wetland soil is alkaline, and natural wetland has stronger alkaline than irrigated wetland. The soluble salt content in artificial irrigated wetland was 0.22%, which is lower than that in natural wetland significantly. The content of soil organic matter (SOM) is the highest in AI wetland, and the lowest in NM. It is because the reed with strong root system and fast growing speed in AI wetland could produce more SOM through microbial decomposition. Other soil nutrients, like total nitrogen (TN), total phosphorus (TP), available nitrogen (N) presented the trend of AI wetland>NE wetland>NM wetland. As the expectation, available phosphorus is a little richer in NM wetland than in NE wetland. Overall, regular artificial irrigation improved water salt distribution pattern and the wetland soil nutrients, so the production of reeds increased.

Table 2 Physicochemical parameters of the soil samples
Notes: Means ± standard errors,*<0.05;**<0.01;***<0.001; ns – not significant. Data followed by the same letter are not significantly different according to the LSD test (<0.05).
3.2 The Statistical Analysis of Sequencing
A total of 364739 raw reads were obtained from the Illimina sequences analysis for the six samples. After tag merge and quality control, a total of 353304 Tags were obtained. After filter sequence number of the low quality and short length, we obtained clean Tags, and effective Tags refers to filter chimeras, eventually for Tags sequence number of the subsequent analysis. After clustering at the 3% cut-off level, a total of 19368 OTUs were achieved. Rare faction curves of soil samples collected in July are steeper than those in September. The rarefaction curves all tend to be flat with sequence number, indicating that most of the bacterial information has been captured. The samples contain much more microbial resources than we expected and require more sequencing effort to reach a saturated stage.
3.3 Soil Bacterial Community Composition
Fig.2 shows the relative abundance of the bacterial phylogenic groups for each wetland site. Sequences that could not be classified into any known group were assigned as unclassified. Groups marked with 1 were samples in July, and the groups marked with 2 were samples in September. All soil samples were dominated by three major bacterial groups (Proteobacteria, Acidobacteria and, Gemmatimonadetes), which made up 75%-80% of the sequences for each sample. Other main phyla include Actinobacteria (2.4%-7.0%), Cyanobacteria (0.04%-6.9%), Bacteroidetes (3.0%-5.6%), Chloroflexi (2.8%-3.9%), Verrucomicrobia (0.8%-2.5%), Planctomycetes (1.1%-2.4%) and Nitrospirae (0.8%-2.3%). The proportion of unclassified sequences varied from 2.5% to 4.4% in the studied soil samples.

Fig.2 Relative abundance of different bacterial phyla among different soils.
Phylum Proteobacteria was the most dominant in all wetland soil groups, and it has been reported previously with wide distribution in wetland (Ahn., 2009; Yu., 2012). The phylum includes a variety of bacterial species related to carbon, nitrogen and phosphorus decomposition, and plays a vital role in wetland material cycling. The abundance of the phylum did not show a clear trend on the whole, but different tendencies were observed on the class level. The content of β-Proteobacte- ria was higher in irragated soils than that in natural wetland soils, which had been reported to have greater ability to decompose cellulose. That may be related to the differences in the composition of soil organic matter. γ-Proteo- bacteria and α-Proteobacteria were more abundant in September soils, nevertheless in samples collected in July β-Proteobacteria and δ-Proteobacteria were the dominant class. The degradation of humic substances mainly rely on some γ-Proteobacteria and α-Proteobacteria, indicating that the soil humification degree is higher in September (Kersters., 2006; Sato., 2009; Rocker., 2012).
In addition to Proteobacteria, Acidobacteria and Gemmatimonadetes are also important composition in soil bacterial community. Acidobacteria (7.8%-26.6%) took greater proportions in irrigated wetland, where organic matter is more abundant and the pH value is lower. The abundance of the Acidobacteria phylum in our study was higher than findings before (Ansola., 2014; Arroyo., 2015). Our result is consistent with findings that the phylum Acidobacteria is often slow growing in the oligotrophic environment. However, Jonesfound that there was a negative correlation between soil pH and the abundance of Acidobacterialassemblage (=−0.80,<0.001) (Jones., 2009). Our study did not find close connection between them as for alkaline soil environment (Fierer., 2007a; Nemergut., 2007). The mechanism of such discrepancy warrants further study.
In irrigated wetland, β-Proteobacteria and acidobacteria were more abundant, which enhanced the degradation of plant residues and the sequestration of organic carbon in the soil. The phylum Gemmatimonadetes was observed in all soils, with slightly higher content in natural wetland than that in irrigated wetland, as a result of low soil moisture in natural wetland (DeBruyn., 2011). It should be noted that the phylum Cyanobacteria has been discovered in all the six samples. In previous study, Cyanobacterium was observed in oil-polluted sites (Al- Hasan., 1998; Abed., 2002). Liaohe oil field is located in our study site, which may result in high abundance of these photosynthetic prokaryotes. Previous research in Liaohe estuarine wetland also confirmed that Cyanobacteria was irreversibly reduced by oil pollution (Tian., 2014). Artificial irrigation increased the soil moisture, and the nutrients such as C, N, P were also input to the soil through irrigated water. Consequently the Acidobacteria, β-Proteobacteria and Gemmatimonadete were more abundant in the irrigated wetland.
In our study, the bacterial community diversity in irrigated wetland is lower than that in natural wetland, suggesting that artificial irrigation increased the production of the reed, while the diversity of bacterial community was decreased. The artificial irrigation increased the flux of soil material input to the wetland and broke the ecological balance in the wetland. The irrigation also caused the anaerobic environment in the soil and suppressed the growth of aerobic bacteria. The bacterial diversity in irrigated wetland is therefore lower than that in natural wetland.
3.4 Diversity of Bacterial Communities
The Shannon-Weaver’s diversity index H’ values in this study were relatively higher than those in other wetlands (Ahn., 2009; Santos., 2011). This is because fertile alluvial and coast delta formed in Liaohe estuary after years of natural and artificial breeding, high content of nutrient elements in soil parent material, and the relatively dry condition in research area is conductive to the growth of a variety of bacteria. H’ varies from 9.09 (AI2) to 9.84 (NM1), The diversity of soil microbial community can be affected by many factors, such as vegetation type, salinity-alkalinity, and hydrological regime (Hu., 2012; Menon., 2013; Peralta., 2013; Urbanová andBárta, 2015). In our study, soil bacterial community diversity in irrigated wetland is lower than that in natural wetland, suggesting that artificial irrigation according to the water requirement rule of reed increased the production of the reeds, while the diversity of bacterium community was decreased. Fixed pattern of artificial irrigation changed the way of wetland soil material input and broke the ecological balance of natural wetlands. Bacterial community is in the anaerobic environment when irrigating, which limits the growth of aerobic bacteria, thus the bacterial diversity is lower. Concerning the impact of season, the soil microbial community diversity is slightly higher in July sample than that in September sample. Additionally, the diversity in NE wetland in July is close to that in September, which may be due to that the reeds need sufficient moisture and nutrients to meet its rapid growth in July. Climate conditions are superior to September, so the bacterial community diversity is higher in July.
3.5 Variation in Bacterial Community with Location and Season
Beta diversity measurements were performed for the soil bacterial community data to examine whether these community patterns differed with location and season.
The shared OTUs for different locations and seasons were determined via the Venn diagram. As shown in Fig.3, a total of 1857 and 1639 OTUs were shared by the communities of the three sampling stations’ in July and September, accounting for 44.91% and 40.58% of the 4135 and 4039 OTUs obtained in total. Some OTUs were only found in a particular bacterial community, as is the case for 384 and 429 bacterial taxa collected in July in both artificial irrigated wetland and natural wetland middle area, which occupied 12.8% and 13.87% of the detected OTUs. Similarly, the bacteria taxa 499 and 431 in September sample covered 18.75% and 14.77% of the detected OTUs, respectively. For example, AI soil involved some eutrophic bacteria, such as,,,, of whichandwere also recorded in Yellow River Estuary (Yu., 2012). Nevertheless, in the NM soils, some halobacteria like,,, andwere discovered, in whichoften appears in reports about extreme environmental microbes, such as the study that detectedin arid soil (Rainey., 2005). However, the shared OTUs among different sampling seasons corresponded to 88.15% and 90.25% of all the OTUs gained, which indicated slight influence of sampling seasons on microbial community. Some studies also found that no seasonal effect is observed in bacterial community structures in the natural and constructed wetlands (He., 2008; Ansola., 2014). While, other studies reported that the content of some species varied in different seasons (Peralta., 2013).

Fig.3 Venn diagram showing the unique and shared OTUs. (A) Venn diagram for three location sets in July; (B) Venn diagram for three location sets in September; (C) Venn diagram for two sets in different months.

Fig.4 Principal coordinates analysis (PCoA) plots derived from weighted Unifrac distances.
Principal coordinates analysis (PCoA) based on weighted Unifrac metric was performed. As shown in Fig.4(A), the sample with high community structure similarity tend to be together, while more difference in microbial community structure may be far apart. The bacterial community composition in wetland soil collected in July tended to be grouped together and was mainly separated from those in September by axis 2. The first two axes explain approximately 72.1% and 15.26% of the variance, respectively, and the third axis is also presented, explaining 6.37% of the cumulative variance. In Fig.4(B), the location effect can be identified. Samples could be grouped by soil collection area. The samples collected in NE and NM soils were closer than those in AI soils. Their distribution adheres to a gradient of soil quality from fertile to poor, in which pH may play an important role, indicating that these two kinds of natural wetlands are similar in soil microbial community structure, while microbial community in wetland with irrigation made some changes.
Soil bacterial community composition is adapt to the environment condition, while other studies have demonstrated that the composition and structure of bacterial communities are often correlated with differences in nutrients, moisture, and pH (Ge., 2008; Kim., 2008; Lauber., 2009). Artificial irrigation affected microbial community structure by taking more nutrients to wetland and reducing the soil pH and salinity. In the future, we need to clarify the mechanism of the effects of various environmental factors on microbial community.
4 Conclusions
Bacterial communities in Liaohe estuarine wetlands were investigated through high-throughput 16S rRNA amplicon sequencing technique. Microbial communities from three different habitats showed obvious differences, which could not be fully explained by the limiting factors determined in present study. The main conclusions are as follows:
All soils were dominated by three major bacterial phylogenic groups (Proteobacteria, Acidobacteria and Gemmatimonadetes). Some eutrophic bacteria were discovered in the irrigated wetland soil, and some halobacteria in natural wetland.
Venn diagram and Beta diversity index heat map indicated that the effect of season on microbial composition and diversity is weaker compared with wetland type. Principal coordinates analysis (PCoA) also revealed that soil bacterial communities can be grouped according to soil nutrient level and sampling seasons. Further study is warranted to fully identify the most critical physicochemical factors controlling the bacterial community structure.
The bacterial communities in irrigated wetland presented lower diversity values, which may be caused by that the fixed pattern of artificial irrigation changed the way of wetland soil material input, broke the ecological balance of natural wetlands, and the metabolic and reproductive differences of bacteria led to lower bacterial diversity.
Acknowledgements
This study was funded by the National Water Pollution Control and Management Technology Major Project of China (No.2013ZX07202-007).
Abed, R. M. M., Safi, N. M. D., Köster, J., de Beer, D., El- Nahhal, Y., and Rullkötter, J., 2002. Microbial diversity of a heavily polluted microbial mat and its community changes following degradation of petroleum compounds., 68: 1674-1683.
Ahn, C., and Peralta, R. M., 2009. Soil bacterial community structure and physicochemical properties in mitigation wetlands created in the Piedmont region of Virginia (USA).,35: 1036-1042.
Ahn, C., Gillevet, P. M., Sikaroodi, M., and Wolf, K. L., 2009. An assessment of soil bacterial community structure and physicochemistry in two microtopographic locations of a palustrine forested wetland., 17: 397- 407.
Al-Hasan, R. H., Al-Bader, D. A., Sorkhoh, N. A., and Radwan, S. S. 1998. Evidence for n-alkane consumption and oxidation by filamentous cyanobacteria from oil-contaminated coasts of the Arabian Gulf., 130: 521-527.
Ansola, G., Arroyo, P., and Miera, L. E. S., 2014. Characterisation of the soil bacterial community structure and composition of natural and constructed wetlands.,473-474: 63-71.
Arroyo, P., Miera, L. E., and Ansola, G., 2015. Influence of environmental variables on the structure and composition of soil bacterial communities in natural and constructed wetlands., 506-507: 380-390.
Barbier, E. B., Hacker, S. D., Kennedy, C., Koch, E. W., Stier, A. C., and Silliman, B. R., 2011. The value of estuarine and coastal ecosystem services., 81: 169-193.
Chao, A., 1987. Estimating the population size for capture- recapture data with unequal catchability., 43: 783- 791.
Clarke, K. R., 1993. Non-parametric multivariate analysis of changes in community structure., 18: 117-143.
DeBruyn, J. M., Nixon, L. T., Fawaz, M. N., Johnson, A. M., and Radosevich, M., 2011. Global biogeography and quantitative season dynamics of gemmatimonadetes in soil., 77: 6295-300.
Dos Santos, H. F., Cury, J. C., do Carmo, F. L., dos Santos, A. L., Tiedje, J., Elsas, J. D., Rosado, A. S., and Peixoto, R. S., 2011. Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: Bacterial proxies for oil pollution., 6: e16943, DOI: 10.1371/journal. pone.0016943.
Edgar, R. C., 2013. UPARSE: Highly accurate OTU sequences from microbial amplicon reads., 10: 996- 998.
Fierer, N., Bradford, M. A., and Jackson, R. B., 2007. Toward an ecological classification of soil bacteria., 88: 1354- 1364.
Ge, Y., Zhang, J., Zhang, L., Yang, M., and He, J., 2008. Long- term fertilization regimes affect bacterial community structure and diversity of an agricultural soil in Northern China., 8: 43-50.
Guittonny-Philippe, A., Masotti, V., Höhener, P., Boudenne, J. L., Viglione, J., and Laffont-Schwob, I., 2014. Constructed wetlands to reduce metal pollution from industrial catchments in aquatic Mediterranean ecosystems: A review to overcome obstacles and suggest potential solutions., 64: 1-16.
He, X. Y., Wang, K. L., Zhang, W., Chen, Z. H., Zhu, Y. G., and Chen, H. S., 2008. Positive correlation between soil bacterial metabolic and plant species diversity and bacterial and fungal diversity in a vegetation succession on Karst., 307: 123-134.
Hu, Y., Li, Y., Wang, L., Tang, Y., Chen, J., Fu, X., Le, Y., and Wu, J., 2012. Variability of soil organic carbon reservation capability between coastal saltmarsh and riverside freshwater wetland in Chongming Dongtan and its microbial mechanism., 24: 1053-1063.
Inceoğlu, O., Al-Soud, W. A., Salles, J. F., Semenov, A. V., and Elsas, J. D., 2011. Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing., 6: e23321, DOI: 10.1371/journal.pone.0023321.
Jones, R. T., Robeson, M. S., Lauber, C. L., Hamady, M., Knight, R., and Fierer, N., 2009. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses.., 3: 442-453.
Kersters, K., De Vos, P., Gillis, M., Swings, J., Vandamme, P., and Stackebrandt, E., 2006. Introduction to the Proteobacteria., 5: 3-37.
Kim, S. Y., Lee, S. H., Freeman, C., Fenner, N., and Kang, H., 2008. Comparative analysis of soil microbial communities and their responses to the short-term drought in bog, fen, and riparian wetlands., 40: 2874- 2880.
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N., 2009. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale., 75: 5111-5120.
Li, H., Bai, J., Yin, N., Zhao, Y. G., and Tian, W. J., 2013. The main factors affecting the number of bacteria in intertidal wetland of the Liaohe Estuary., 43: 67-71 (in Chinese with English abstract).
Ligi, T., Oopkaup, K., Truu, M., Preem, J. K., Nãlvak, H., Mitsch, W. J., Mander, Ü., and Truu, J., 2013. Characterization of bacterial communities in soil and sediment of a created riverine wetland complex using high-throughput 16S rRNA amplicon sequencing., http:// dx.doi.org/10.1016/ j.ecoleng.2013.09.007.
López-Piñeiro, A., Muñoz, A., Zamora, E., and Ramírez, M., 2013. Influence of the management regime and phenological state of the vines on the physicochemical properties and the seasonal fluctuations of the microorganisms in a vineyard soil under semi-arid conditions., 126: 119-126.
Lu, R. K., 2000.. China’s Agricultural Science and Technology Press, Beijing, 1-44.
McCune, B., and Grace, J. B., 2002. Analysis of ecological communities. MJM software design, Gleneden Beach, Oregon, http://www.pcord.com.
Menon, R., Jackson, C. R., and Holland, M. M., 2013. The influence of vegetation on microbial enzyme activity and bacterial community structure in freshwater constructed wetland sediments., 33: 365-378.
Nemergut, D. R., Anderson, S. P., Cleveland, C. C., Martin, A. P., Miller, A. E., Seimon, A., and Schmidt, S. K., 2007. Microbial community succession in an unvegetated, recently deglaciated soil., 53: 110-122.
Phillips, L. A., Schefe, C. R., Fridman, M., O’Halloran, N., Armstrong, R. D., and Mele, P. M., 2015. Organic nitrogen cycling microbial communities are abundant in a dry Australian agricultural soil., 86: 201- 211.
Pereira, E., Silva, M. C., Semenov, A. V., Schmitt, H., Elsas, J. D., and Salles, J. F., 2013. Microbe-mediated processes as indicators to establish the normal operating range of soil functioning., 57: 995-1002.
Peralta, A. L., Ludmer, S., Matthews, J. W., and Kent, A. D., 2014. Bacterial community response to changes in soil redox potential along a moisture gradient in restored wetlands., 73: 246-253.
Peralta, A. L., Ludmer, S., and Kent, A. D., 2013. Hydrologic history influences microbial community composition and nitrogen cycling under experimental drying/wetting treatments., 66: 29-37.
Peralta, R. M., Ahn, C., and Gillevet, P. M., 2013. Characterization of soil bacterial community structure and physicochemical properties in created and natural wetlands., 443: 725-732.
Peng, R., Zou, L., and Wan, H., 2012. Studies on the accumulation of organic carbon in the soil in the reed wetland at Liaohe Estuary., 42 (5): 28-34.
Rainey, F. A., Ray, K., Ferreira, M., Gatz, B. Z., Nobre, M. F., Bagaley, D., Rash, B. A., Park, M. J., Earl, A. M., Shank, N. C., Small, A. M., Henk, M. C., Battista, J. R., Kämpfer, P., and Costa, M. S., 2005. Extensive diversity of ionizing-ra- diation-resistant bacteria recovered from Sonoran Desert soil and description of nine new species of the genus Deinococcus obtained from a single soil sample., 71: 5225-5235.
Rocker, D., Brinkhoff, T., Grüner, N., Dogs, M., Simon, M., 2012. Composition of humic acid-degrading estuarine and marine bacterial communities., 80: 45- 63.
Roesch, L. F., Fulthorpe, R. R., Riva, A., Casella, G., Hadwin, A. K. M., Kent, A. D., Daroub, S. H., Camargo, F. A. O., Farmerie, W. G., and Triplett, E. W., 2007. Pyrosequencing enumerates and contrasts soil microbial diversity., 4: 283- 290.
Sato, K., Kato, Y., Taguchi, G., Nogawa, M., Yokota, A., and Shimosaka, M., 2009. Chitiniphilus shinanonensis gen. nov., sp. nov., a novel chitin-degrading bacterium belonging tobacteria., 55: 147-153.
Suleiman, A. K., Manoelib, L., Boldo, J. T., Pereira, M. G., and Roesch, L. F. W., 2013. Shifts in soil bacterial community after eight years of land-use change., 36: 137-144.
Tian, W., Zhao, Y., Sun, H., Bai, J., Wang, Y., and Wu, C., 2014. The effect of irrigation with oil-polluted water on microbial communities in estuarine reed rhizosphere soils., 70: 275-281.
Urbanová, Z., and Bárta, J., 2015. Effects of long-term drainage on microbial community composition vary between peatland types., 92: 16-26.
Wardle, D. A., Bardgett, R. D., Klironomos, J. N., Setälä, H., Putten, W. H., and Wall, D. H., 2004. Ecological linkages between aboveground and belowground biota., 304: 1629-1633.
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R., 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy., 73: 5261-5267.
Wang, Y., Sheng, H. F., He, Y., Wu, J. Y., Jiang, Y. X., Tam, N. F., and Zhou, H. W., 2012. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of Illumina tags., 78: 8264-8271.
Wang, Z. Y., Xin, Y. Z., Gao, D. M., Li, F. M., Morgan, J., and Xin, B. S., 2010. Microbial community characteristics in a degraded wetland of the Yellow River Delta., 20: 466-478.
Whittaker, R. H., 1972. Evolution and measurement of species diversity., 21: 213-251.
Xu, Z., Yu, G., Zhang, X., Ge, J., He, N., Wang, Q., and Wang, D., 2014. The variations in soil microbial communities, enzyme activities and their relationships with soil organic matter decomposition along the northern slope of Changbai Mountain.,86: 19-29.
Yin, N., Wang, L. P., He, S. L., and Bai, J., 2014. Characteristics of soil microbial community structure in the process of ecological restoration of Liaohe estuary wetland., 38: 195- 199 (in Chinese with English abstract).
Yu, Y., Wang, H., Liu, J., Wang, Q., Shen, T., Guo, W., and Wang, R., 2012. Shifts in microbial community function and structure along the successional gradient of coastal wetlands in Yellow River Estuary., 49: 12-21.
Zhang, X., Wang, Z., Liu, X., Hu, X., Liang, X., and Hu, Y., 2013. Degradation of diesel pollutants in Huangpu-Yangtze
River estuary wetland using plant-microbe systems., 76: 71-75.
Zhang, Y., Zheng, X., and Wu, C., 2011. Experimental study of evapotranspiration from phragmites australis wetland in Liaohe Estuary., 22 (3): 352-358.
Zhao, X. L., Zhou, G. S., and Lu, G. H., 2009. The study on soil microbial characteristic under different types of vegetation in Liaohe Delta., 40: 1266-1269 (in Chinese with English abstract).
Zhao, X. L., Zhou, G. S., Zhou, L., Lu, G. H., Jia, Q. Y., and Xie, Y. B., 2008. Seasonal changes in soil microbial biomass C in bulrush wetlands of Panjin, Northeast China., 39: 43-46 (in Chinese with English abstract).
(Edited by Ji Dechun)
(Received March 9, 2017; revised September 15, 2017; accepted November 22, 2017)
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2018
. E-mail: hhu@ouc.edu.cnE-mail: zhengyan@ouc.edu.cn
杂志排行

Journal of Ocean University of China的其它文章
- Offshore Fault Geometrics in the Pearl River Estuary, Southeastern China: Evidence from Seismic Reflection Data
- Application of Geoid Anomalies to the Tectonic Research in the East Asian Continental Margin
- Middle Holocene Organic Carbon and Biomarker Records from the South Yellow Sea: Relationship to the East Asian Monsoon
- Mesozoic Deformation and Its Geological Significance in the Southern Margin of the South China Sea
- Optimization of Shanghai Marine Environmental Monitoring Sites in the Identification of Boundaries of Different Water Quality Grades
- Seasonal Variation of Environmental Variables and Phytoplankton Community Structure and Their Relationship in Liaodong Bay of Bohai Sea, China