HPLC法测定盐酸莫西沙星氯化钠注射液中脱羧莫西沙星的含量
2018-08-10于黎鑫刘玉玉
王 鹏 于黎鑫 刘玉玉
(山东齐都药业有限公司,山东淄博255400)
0 引言
盐酸莫西沙星化学名为1-环丙基-7-{(S,S)-2,8-重氮-二环[4.3.0]壬-8-基}-6-氟-8-甲氧-1,4-二氢-4-氧-3-喹啉羧酸盐酸盐,是由德国拜耳公司研制的第4代氟喹诺酮类超广谱抗生素,它保留了第3代抗革兰阴性菌的活性,其8-甲氧基部分还提高了抗革兰阳性菌的活性,特别是增加了对厌氧菌的抗菌活性,是具有抗菌活性的广谱抗菌药。莫西沙星的耐药性使得对糖肽类、头孢菌素类、青霉素类、四环素类和大环内酯类耐药的机制不会影响莫西沙星的抗菌活性,莫西沙星与这些抗菌药无交叉耐药性,且至今未发现质粒介导的耐药性出现。
现行版《美国药典》中盐酸莫西沙星的有关物质检查采用ODS柱,用甲醇-磷酸盐系统进行梯度洗脱,仅对杂质A、B、C、D、E等5个已知杂质进行了控制。
《欧洲药典》9.0版盐酸莫西沙星的有关物质检查采用的色谱柱为苯基柱,以甲醇-四丁基硫酸氢铵磷酸盐缓冲液为流动相,同样仅对上述5个已知杂质进行了控制。
盐酸莫西沙星国内现行标准(YBH01912013)中的有关物质检查也采用了《欧洲药典》9.0版盐酸莫西沙星有关物质检查的方法,对上述5个杂质以及杂质F进行了控制。
现根据盐酸莫西沙星的合成工艺,对杂质谱进行解析,除了对可能存在的上述6个杂质进行研究外,新增了对降解杂质脱羧莫西沙星的研究,其化学结构式如图1所示,提高了盐酸莫西沙星氯化钠注射液质量标准。
采用上述盐酸莫西沙星有关物质检测方法及多篇文献报道的检测方法进行测试,均不能有效测定脱羧莫西沙星含量。最终,通过参考盐酸莫西沙星氯化钠注射液进口药品注册标准(JX20030036),对标准中的有关物质检查方法进行了优化,从而建立了盐酸莫西沙星中脱羧莫西沙星的含量测定方法,并经过详细的方法学验证,证明了所建立的方法准确度高、重复性好,可以用来测定盐酸莫西沙星氯化钠注射液中脱羧莫西沙星的含量。
1 仪器与试药
1.1 仪器
LC-20A型高效液相色谱仪,日本岛津公司;CPA225D型电子天平,北京赛多利斯仪器系统有限公司;FE20型pH计,梅特勒-托利多公司。
1.2 试药
杂质A对照品(批号:XM56-1023,纯度99.90%)、杂质C对照品(批号:XM56-6738,纯度99.13%)、杂质F对照品(批号:XM56-5520,纯度99.88%),均来自HI-CHEMICAL;杂质B对照品(批号:17-M00022-05,纯度99.2%)、杂质D对照品(批号:16-M00024-02,纯度99.3%)、杂质E对照品(批号:15-M00025-12,纯度97.5%),均来自RXA;脱羧莫西沙星对照品(批号:1672226M-HB-08,纯度99.1%),深圳斯坦德化工科技有限公司;盐酸莫西沙星氯化钠注射液(批号:17092901、17092902、17092903),山东齐都药业有限公司;甲醇为色谱纯,水为纯化水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件与系统适用性

图1 盐酸莫西沙星及脱羧莫西沙星化学结构式
色谱柱:菲罗门Gemini-NX C18色谱柱(4.6 mm×250 mm,5μm);流动相:1%三乙胺溶液(用磷酸调节pH值至2.5)∶甲醇=70∶30;检测波长:280 nm;流速:1.0 mL/min;柱温:40 ℃;进样量:10 μL。理论板数以脱羧莫西沙星峰计应不低于2 000。
2.2 溶液的制备
(1)溶剂:取无水亚硫酸钠约0.050 g,加水约500mL使其溶解,加入磷酸1mL,用水稀释至1000mL。
(2)空白辅料溶液:按盐酸莫西沙星氯化钠注射液处方配制不含盐酸莫西沙星的溶液,灭菌,作为空白辅料;再用溶剂稀释两倍,作为空白辅料溶液。
(3)对照品溶液:分别取杂质A、B、C、D、E、F与脱羧莫西沙星对照品适量,精密称定,用溶剂制成每1 mL中含各杂质16μg的溶液,作为对照品贮备液;从中精密量取1 mL,置于20 mL容量瓶中,用溶剂定容至刻度,摇匀,即得。
(4)系统适用性溶液:精密量取本品10 mL与对照品贮备液1 mL,置于20 mL容量瓶中,用溶剂稀释至刻度,摇匀,即得。
(5)供试品溶液:精密量取本品10 mL,置于20 mL容量瓶中,用溶剂稀释至刻度,摇匀,即得。
2.3 专属性考察
分别精密量取空白辅料溶液、对照品溶液、系统适用性溶液各10μL,注入液相色谱仪,记录色谱图,结果如图2所示。空白辅料不干扰脱羧莫西沙星的含量测定,脱羧莫西沙星峰与相邻峰分离度良好。
结果表明:所建立的高效液相色谱法的专属性良好。
2.4 强制降解试验
取盐酸莫西沙星氯化钠注射液(批号:17092901)适量,分别考察在酸、碱、氧化、高温、光照条件下样品的质量稳定情况,具体条件如表1所示。
试验结果表明:空白溶液不干扰脱羧莫西沙星含量的测定。盐酸莫西沙星氯化钠注射液在酸、碱、高温条件下,脱羧莫西沙星含量均有所升高;在氧化条件下,脱羧莫西沙星含量有所下降;在光照条件下,脱羧莫西沙星含量几乎不变。
2.5 系统适用性试验
取系统适用性溶液,进样6次,记录色谱图。从色谱图中可以看出,脱羧莫西沙星峰与相邻峰之间的分离度及其理论板数、拖尾因子均符合要求。计算得到脱羧莫西沙星的峰面积RSD为0.68%。
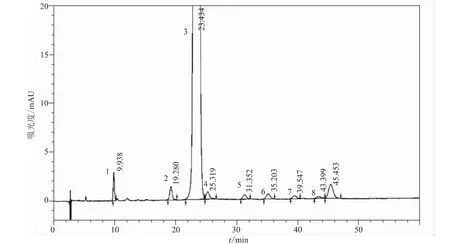
图2 专属性试验液相色谱图

表1 强制降解试验条件
结果表明:所建立的方法系统适用性良好。
2.6 检测限和定量限
精密称取脱羧莫西沙星对照品适量,用溶剂溶解并依次稀释,分别取10μL注入液相色谱仪,记录色谱图。
检测限信噪比S/N=3,定量限信噪比S/N=10。脱羧莫西沙星的检测限为0.065 2μg/mL,定量限为0.217 5μg/mL。
2.7 线性关系考察
精密称取脱羧莫西沙星对照品适量,用溶剂溶解并分别制成定量限浓度不低于150%限度范围内的系列线性溶液。
分别精密量取10μL,注入液相色谱仪,记录色谱图。以浓度为横坐标,峰面积为纵坐标,绘制标准曲线,得到线性回归方程Y=42913X-1518.3(r=0.9985)。
结果表明:脱羧莫西沙星在0.2175~1.0875μg/mL进样浓度范围内,线性关系良好。
2.8 重复性试验
按拟定的方法配制对照品溶液,再平行配制6份系统适用性溶液(作为重复性试验供试品溶液1~6)。分别取上述溶液10μL,依法进样,记录色谱图,按外标法以峰面积计算脱羧莫西沙星的含量。6次测量结果显示:脱羧莫西沙星的含量平均值为0.11%,6次结果的RSD为0.01%。
结果表明:所建立的方法重复性良好。
2.9 加样回收率试验
取“重复性试验”项下的对照品溶液,再配制含脱羧莫西沙星限度的80%、100%、120%3个不同浓度的供试品溶液各3份。分别取上述溶液10μL,依法进样,记录色谱图,计算脱羧莫西沙星的回收率结果,如表2所示。
结果表明:平均回收率为102.79%,RSD为1.62%。
2.10 耐用性考察
2.10.1 溶液稳定性
按拟定的方法配制对照品溶液及系统适用性溶液(作为溶液稳定性试验的供试品溶液),在室温下放置24 h,分别于0 h、3 h、6 h、9 h、12 h、18 h、24 h精密量取10μL,注入液相色谱仪,记录色谱图。
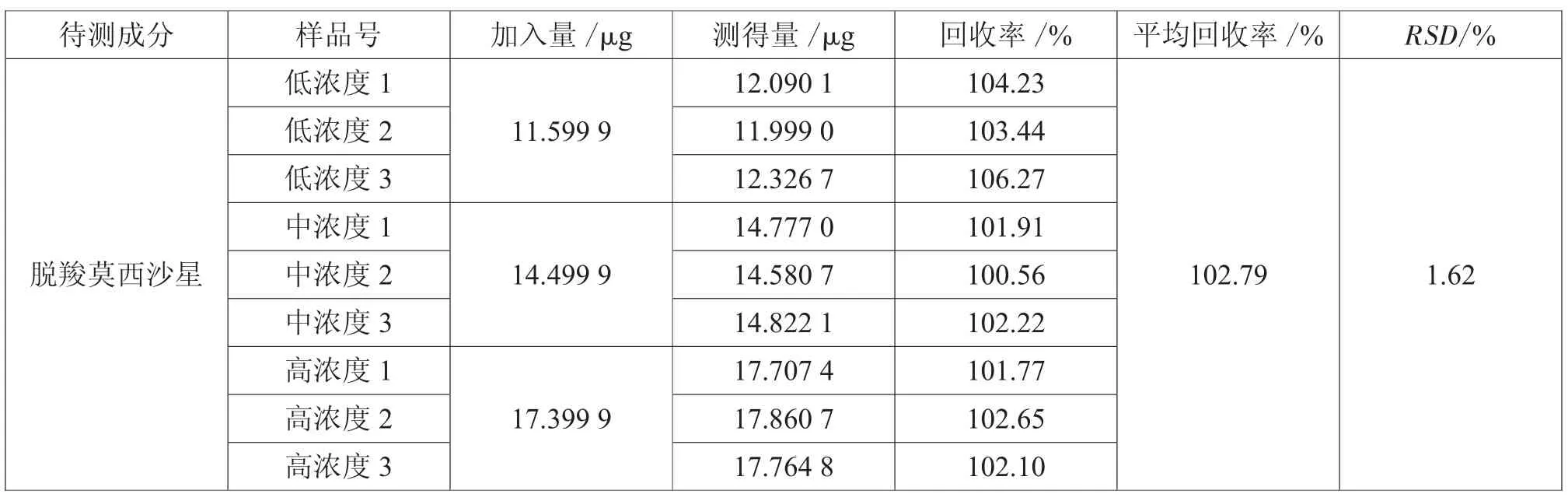
表2 加样回收率试验
结果表明:对照品溶液与供试品溶液在室温条件下放置24 h,脱羧莫西沙星峰面积的RSD分别为1.42%、0.79%,表明溶液稳定性良好。
2.10.2 色谱条件参数变化
将色谱条件参数柱温(±3℃)、流速(±0.1mL/min)、流动相比例(±2%)、流动相pH(±0.2)、色谱柱(不同批次)等进行微小的变动,以此评估其对脱羧莫西沙星含量测定结果的影响程度。
结果表明:当参数发生微小变动时,脱羧莫西沙星的含量测定结果几乎不受影响,表明所建立的方法耐用性较好。
2.11 样品测定
取中试规模的盐酸莫西沙星氯化钠注射液样品3批(批号:17092901、17092902、17092903),依法测定脱羧莫西沙星含量,结果如表3所示。
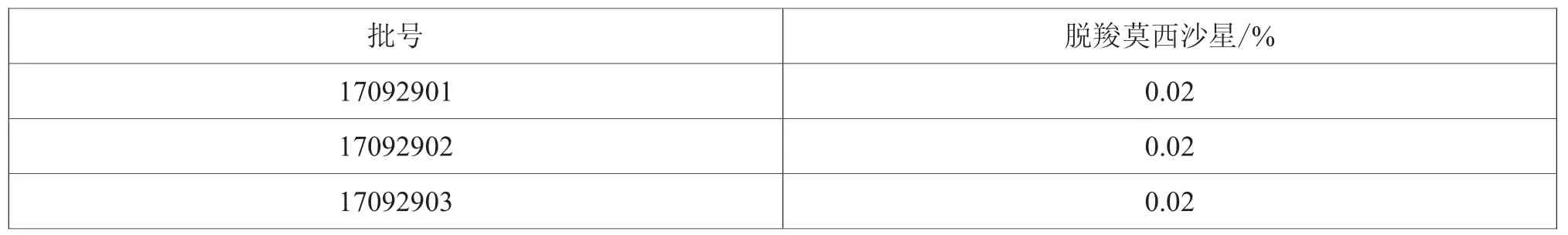
表3 样品测定脱羧莫西沙星含量结果
3 结语
本文根据盐酸莫西沙星杂质谱的解析,除对相关文献已报道的杂质A、B、C、D、E、F进行控制外,新增了对降解杂质脱羧莫西沙星的研究,提高了盐酸莫西沙星氯化钠注射液的质量标准。参考盐酸莫西沙星氯化钠注射液进口药品注册标准而建立的盐酸莫西沙星中脱羧莫西沙星的含量测定方法,经详细全面的方法学研究证明,该方法测定的结果准确,重复性良好,可以用来测定盐酸莫西沙星氯化钠注射液中脱羧莫西沙星的含量。
