Various changes in cryopreserved acellular nerve allografts at −80° C
2018-08-09YanYanHuangXiaoLuXuXiJunHuangJiangHuiLiuJianQiShuangZhuZhaoWeiZhuBoHeQingTangZhuYangBinXuLiQiangGuXiaoLinLiu
Yan-Yan Huang, Xiao-Lu Xu, Xi-Jun Huang, Jiang-Hui Liu, , Jian Qi, Shuang Zhu, Zhao-Wei Zhu, , Bo He, , Qing-Tang Zhu,Yang-Bin Xu, Li-Qiang Gu, Xiao-Lin Liu
1 Department of Plastic Surgery, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong Province, China
2 Yale School of Medicine, New Haven, CT, USA
3 Department of Orthopedic and Microsurgery, the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong Province, China
4 Department of Emergency, the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong Province, China
5 Department of Joint and Orthopedic, Orthopedic Center, Zhujiang Hospital of Southern Medical University, Guangzhou, Guangdong Province,China
Abstract The experimental design evaluated histological, mechanical, and biological properties of allogeneic decellularized nerves after cryopreservation in a multi-angle, multi-directional manner to provide evidence for long-term preservation. Acellular nerve allografts from human and rats were cryopreserved in a cryoprotectant (10% fetal bovine serum, 10% dimethyl sulfoxide, and 5% sucrose in RPMI1640 medium) at −80° C for 1 year, followed by thawing at 40° C or 37° C for 8 minutes. The breaking force of acellular nerve allografts was measured using a tensile test.Cell survival was determined using L-929 cell suspensions. Acellular nerve allografts were transplanted into a rat model with loss of a 15-mm segment of the left sciatic nerve. Immunohistochemistry staining was used to measure neurofilament 200 expression. Hematoxylin-eosin staining was utilized to detect relative muscle area in gastrocnemius muscle. Electron microscopy was applied to observe changes in allograft ultrastructure. There was no obvious change in morphological appearance or ultrastructure, breaking force, or cytotoxicity of human acellular nerve allografts after cryopreservation at −80° C. Moreover, there was no remarkable change in neurofilament 200 expression, myelin sheath thickness, or muscle atrophy when fresh or cryopreserved rat acellular nerve allografts were applied to repair nerve injury in rats. These results suggest that cryopreservation can greatly extend the storage duration of acellular nerve tissue allografts without concomitant alteration of the physiochemical and biological properties of the engineered tissue to be used for transplantation.
Key Words: nerve regeneration; acellular nerve allografts; cryopreservation; storage; transplantation; nerve; neural regeneration
Introduction
Increasing incidence of peripheral nerve damage has been observed as a result of various injuries (World Health Organization, 2000; He et al., 2014). Technological developments have prompted regenerative medicine to replace the methodology of“healing a wound with a wound” for the repair of nerves. In particular, acellular tissue matrix is being widely investigated as a novel approach to organ repair and tissue regeneration and is being actively explored as a bio-derivative (Badylak et al., 2009;Ozbek et al., 2010; Zhu et al., 2015; Xiang et al., 2017). Following clinical application of an acellular allogeneic nerve graft (ANA)(Avance, AxoGenW, Alachua, FL, USA), we generated an allogeneic acellular tissue matrix for nerve repair in May 2012 and obtained a drug and medical device registration certificate from the China Food and Drug Administration (license number:CFDSM2012-3460641) (He et al., 2015; Tang et al., 2017). This makes China the second country in the world to systematically understand the engineering behind decellularized peripheral nerve scaffolds and successfully apply these scaffolds in cases of peripheral nerve damage and repair in a clinical setting. This represents a major milestone in biomaterials research for both tissue engineering and regenerative medicine fields.
In our earlier studies, decellularized nerve tissues were grafted immediately after generation, and therefore, long-term preservation of biomaterials was not needed. However, in order for acellular tissue matrix to be put into broad commercial use, long-term preservation of these biomaterials is crucial for tissue collection, production, sale, distribution, and usage.Thus, new methods capable of extending the shelf life of acellular nerve allografts are urgently needed. Avance®processed allograft is preserved by dry ice; however, the details of this preservation method remain unknown. In 1949, the English biologist Dr. Christopher Polge discovered that glycerol-treated sperm could survive for a prolonged period of time under hypothermic conditions without cell death, and successfully applied this method in a clinical setting (Polge et al., 1949).Since then, use of cryopreservation has significantly progressed and been widely adopted by others. Wang et al. (2002) recovered rat ovaries following cryopreservation and successfully transplanted them into syngeneic recipients after thawing. In 2014, we reported that rat hind limbs can be cryopreserved and re-implanted into rats with restored blood supply (Wang et al.,2014). Therefore, we applied cryopreservation to ANA storage and hypothesized that deep low temperature might have no obvious negative effects. Thus, the purpose of this study was to understand the effects of cryopreservation on the histological,mechanical, and biological properties of ANAs.
Materials and Methods
In vitro experiments using human acellular nerve allograft(hANA)Preparation of hANA
hANAs were prepared according to a previously described method (Yang et al., 2011). Briefly, fresh hANAs were obtained from Zhongshan University Medical Equipment (Guangzhou,China; license number: CFDSM2012-3460641). Nerves with a diameter of 6 mm were chosen for this study. Immediately after harvest, nerves were rinsed three times in chilled deionized water to remove contaminating blood, fat, and connective tissues from the epineurium using a surgical microscope. Nerves were then cut into segments 40–50 mm in length.
A modified protocol based on a previous method (Sondell et al., 1998) was used to prepare nerve scaffolds. Briefly, nerves were agitated in deionized distilled water for 6 hours before replacing the water with 46 mM Triton X-100 (in distilled water). After 24 hours of agitation, scaffolds were rinsed three times with 10 mM phosphate-buffered saline (PBS) solution(10 minutes per wash). Nerves were then agitated for an additional 24 hours in deionized distilled water containing 96 mM sodium deoxycholate. The above steps were repeated before a final wash of nerves in 10 mM PBS solution. The ratio of tissue to solution was 1:100 (v/v). All treatment steps were performed at room temperature.
Cryopreservation
Following the rinsing process, twenty grafts were randomly selected and divided into non-cryopreservation and cryopreservation groups. Grafts from the non-cryopreservation group were stored in 10 mM PBS and analyzed immediately, while grafts in the cryopreservation group were maintained in a cryoprotectant containing 10% fetal bovine serum, 10% dimethyl sulfoxide (DMSO), and 5% sucrose in RPMI1640 medium at−80° C for 1 year.
Thawing process
A total of 200 mL of eluent RPMI1640 medium (Gibco, Invitrogen, Carlsbad, CA, USA) was placed in a sterile beaker in a 40° C water bath. After the eluent reached 40° C, frozen nerves were quickly removed from liquid nitrogen storage and placed into the eluent for eight minutes.
Evaluation of mechanical properties
Mechanical properties of nerve allografts were compared in different groups using mechanical testing equipment for biomaterials. The breaking force (tensile strength) was measured using a tensile test with Benchtop Testers (H1K-S UTM, Tinius Olsen, TMC, PA, USA). For each test, the equipment applied a drawing speed of 10 mm/min.
Cell toxicity test
First, 0.96 g of acellular nerve graft was placed into 4.8 mL of PBS at a ratio of 0.2 g/mL and incubated for 24 hours at 37± 2°C. Suspensions of L-929 cells (Cellcook, Guangzhou,China) were prepared at a density of 2.5 × 105cells/mL. Cells were then seeded into a 96-well plate and incubated for 24 hours in an incubator with a stable temperature and 5% CO2.When cells grew to confluency in a monolayer, the medium was changed. Fresh cell medium was added into wells in the control group, while previously prepared acellular extracts(with serum) were added to wells of the experimental group and placed back at 37°C in a CO2incubator for an additional 24 hours. Cell morphology was monitored under a BX60 microscope (Olympus, Tokyo, Japan) as 20 µL of 5 g/L 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide solution was added into each well and incubated for 4 hours. After incubation, solutions from each well were removed and 200 µL of DMSO was added. The absorbance at 570 nm was then recorded. Relative growth rates of cells were then calculated based on the reading. Based on the relative growth rate, cell toxicity was graded as 0 (relative growth rate≥ 100%), I (75–99%), II (50–74%), III (25–49%), IV(1–24%) or V (0%). Of these, grades I and II were recognized as acceptable, whereas grades III–V were considered unacceptable.
Scanning electron microscopy
Scanning electron microscopy was used to observe the ultrastructure of hANAs. Samples were prepared according to our previous study (Zhu et al., 2016). Briefly, samples were ultrathin sectioned after freeze-drying treatment and the surface of the slice was sputter-coated with gold. Samples were imaged using a JSM-7600F scanning electron microscope (JEOL, Tokyo,Japan). Images from six random fields of each section were analyzed using Image-Pro Plus (Version 6.0).
In vivo experiments using rat acellular nerve allograft (rANA)Preparation of rANA
Forty-nine clean healthy male Sprague-Dawley rats aged 3 months old and weighing 180–250 g were selected as donors of nerve allografts. Rats were purchased from the Animal Center of the First Affiliated Hospital of Sun Yat-sen University of China (license number: SYXK (Yue) 2012-0081). Thirteen rats were intraperitoneally anesthetized with 10% chloral hydrate(0.3 mL/100 g body weight) (Sinopharm Chemical Reagent Co., Shanghai, China). Bilateral sciatic nerves (≥ 20 mm in length) were excised from rats, cleaned of external fat and connective tissue, and immediately placed in sterile PBS. Nerve segments were treated with the same procedure as described above for hANA. This study was approved by the Experimental Animal Administration Committee of Sun Yat-Sen University of China (approval number: [2013]A-055). Efforts were undertaken to minimize animal suffering during experiments.
rANA transplantation
Thirty-six healthy male Sprague-Dawley rats were randomly divided into three groups: (1) normal group without any surgery (n = 12), (2) non-cryopreservation group (n = 12), and(3) cryopreservation group (n = 12). At first, all rats were intraperitoneally anesthetized with 10% chloral hydrate (10 mL/kg). Procedures were as described in a previous publication(Zhu et al., 2015). Under aseptic conditions, the skin of the left leg was cut parallel to the femur, and the sciatic nerve was exposed by splitting the superficial gluteus muscle. Under a surgical microscope, a 15-mm segment of the left sciatic nerve was severed and removed near the obturator tendon in the midthigh (Pan et al., 2009; Wilson et al., 2010). In the non-cryopreservation group, a 15-mm segment of the sciatic nerve was removed and replaced with an 18-mm donor rANA previously stored at 4°C for no more than one week. In the cryopreservation group, an 18-mm donor rANA previously stored in cryoprotectant at −80° C for 1 year (thawed at 37° C) was transplanted into the niche inside the recipient. The muscle and skin were then connected with 6-0 and 4-0 polyamide sutures, respectively.
Immunohistochemistry
On day 28 after surgery, six rats from each group were randomly selected and sacrificed to harvest nerve grafts (n = 6).All nerves were fixed in 4% paraformaldehyde for 24 hours at 4°C, followed by dehydration in 20% sucrose solution for 24 hours and a second dehydration in 30% sucrose solution for an additional 24 hours. The middle portion of the nerve was cut into 10-μm-thick sections using a cryostat. These sections were then mounted on slides and subjected to immunohistochemistry with a rabbit anti-rat neurofilament 200(NF-200) antibody (1:400; Sigma-Aldrich, Tokyo, Japan) and goat anti-rabbit secondary antibody conjugated to fluorescein isothiocyanate (1:200; TCS Biologicals, Botolph Clayton, UK).Antibodies were diluted in PBS solution containing 3% rat serum, 3% goat serum, and 0.02% sodium azide (Sigma-Aldrich)to reduce background staining.
Computer-assisted imaging analysis (Seescan Analytical Services, Cambridge, UK) was used to identify regenerating axons and blood vessels within grafts. The middle portions of grafts were examined. The integrated optical density of positive staining was assessed using Image-Pro Plus 6.0 (Media Cybernetics,Rockville, MD, USA). Three random sections were selected from each rat for analysis and the results were averaged.
Hematoxylin-eosin staining
At 28 days after surgery, six rats in each group were randomly selected concomitant to nerve harvesting. Gastrocnemius muscles were collected for hematoxylin-eosin staining. Muscle samples were fixed in paraformaldehyde, embedded in paraffin, and sectioned in a transverse manner. Sections were then stained with hematoxylin and eosin. For each sample, images were obtained from five random fields. Image-Pro Plus (Version 6.0) software was used to measure muscle area. Five samples from each group were used for statistical analysis. Relative muscle area was calculated by normalizing the value to the contralateral normal muscle.
Electron microscopy
On day 28 after surgery, six rats from each group were randomly selected and sacrificed to harvest nerve grafts (n = 6) for electron microscopy. Transmission electron microscopy was used to estimate myelin sheath regeneration. Ultrathin sections were stained with lead citrate and uranyl acetate, and then examined under a transmission electron microscope (CM120;Philips Amsterdam, Netherlands) equipped with an image-acquisition system that was used to measure the thickness of myelin sheaths according to a previous study (Zhou et al., 2014). Images from six random fields of each section were analyzed using Image-Pro Plus software (Version 6.0).
Statistical analysis
Data are presented as a percentage or as the mean ± SD. Statistical analysis was performed using SPSS 13.0 software (SPSS,Chicago, IL, USA). Comparisons between different groups were performed using analysis of variance. A value of P < 0.05 was defined as the threshold for statistical significance.
Results
In vitro experimental results of hANA
hANA general morphological assessment
All nerve samples manifested as a milky strip under natural light (Figure 1A, B).
hANA histological features
Under the microscope, transverse sections of ANAs demonstrated preservation of both the epineurium and perineurium with a significant amount of extracellular matrix, but no cellular components were observed after chemical extraction.Endoneurial tubes resembled “empty bubbles” within the tissue, regardless of whether grafts were preserved under normal temperature or cryopreservation, and no significant morphological difference was observed in nerves before and after cryopreservation (Figure 1C, D).
Ultrastructural change of hANA
Nerves demonstrated preservation of gross structure by scanning electron microscopy, as characterized by ordered, weblike empty tubes surrounded by dense fibers and the absence of axons and myelin sheaths. Similarly, no apparent differences were observed between nerve allografts preserved under the two different conditions (Figure 1E, F).
Mechanical properties of hANA
No statistically significant difference was observed in the mechanical measurements of nerve grafts in non-cryopreservation and cryopreservation groups, and their tensile strengths were 18.2 ± 2.7 N, vs. 17.1 ± 3.2 N, respectively (P > 0.05).
hANA cytotoxicity
After L-929 cells were incubated with either fresh or acellular extracts for 48 hours, the relative growth rate was determined to be grade I for both groups, suggesting that cryopreservation did not induce cell toxicity.
In vivo experiment results of rANA Axonal growth of transplant rANA in rats by immunofluorescence
Twenty-eight days after surgery, the extent of axonal regeneration was evaluated by immunostaining for NF-200 in longitudinal sections of nerve allografts from both treatment groups(Figure 2N1–N4). Compared with normal sciatic nerves,there were differences in both groups. However, it was apparent from these stains that the NF-200 expression level (analysis of integrated optical density) of nerve grafts preserved in conditions of normothermia was not significantly different from the level in cryopreserved samples (116,201.90 ± 5443.15 versus 121,802.60 ± 4805.15; P > 0.05).
Histological properties of gastrocnemius muscles in rats after rANA transplantation
The extent of muscle atrophy was assessed 28 days after surgical removal of nerves in rats (Figure 2M1–M4). Compared with healthy gastrocnemius muscles on the non-surgical side of the limb, no significant difference was observed in stained cross-sections of rANA muscle fibers between the two preservation methods (59.89 ± 3.78% versus 62.78 ± 8.10%; P >0.05; Figure 2).
Myelin sheath changes in rat sciatic nerves after rANA transplantation
Twenty-eight days after surgical removal of sciatic nerves,myelin regeneration was measured by transmission electron microscopy (Figure 2T1–T4). The results indicated that cryopreserved nerve grafts contained myelinated nerves with structure, morphology, and distribution similar to grafts that were not preserved at an extremely low temperature. In addition,although differences could be seen between groups compared with normal sciatic nerves, statistically significant differences were not observed in myelin sheath thickness between the two groups (4.48 ± 1.70 μm versus 4.50 ± 0.89 μm; P > 0.05;Figure 2).
Discussion
In this study, acellular nerve biomaterials were stored at 4°C after sterilization. However, this tissue may only be stored by this method for a limited period. Numerous studies have suggested that cryopreservation can reduce the metabolic activity of cells and delay functional loss. In a clinical setting, biological tissues and organs are often cryopreserved using special methods and are maintained for prolonged periods; they are only thawed to normal temperatures to recover their function when needed (He and Wang, 2007). The well-known Arrhenius equation forms the mathematical basis of our study. As shown below, A is the Arrhenius factor, the value of which is constant and dependent upon a particular reaction. In this equation,cell and tissue activity is a function of temperature (T) (Wang et al., 2014). Theoretically, any enzymatic or chemical reaction that damages the cell could cease when the temperature drops to a certain degree, which would indefinitely prolong the preservation of tissue or cells. In practice, most needs for longterm preservation can be met when the tissue temperature is as low as the typical temperature of liquid nitrogen (Wang et al.,2014).
Based on the formula κ = A × exp(−Bα/RT), where κ is the reaction rate constant; R is the molar gas constant; Eα is the apparent activation energy; and A is the pre-exponential factor (also called frequency factor), cells can be preserved for dozens of hours at 4° C, for several days at −40° C, for several months at −80° C and for centuries at −196° C (He and Wang, 2007; He et al., 2010). The lower the preservation temperature, the longer tissue can be stored. For practical use,−196° C would fulfill most of our tissue preservation needs.Therefore, it is predicted that ANAs may be stored for decades or even centuries; although, our study only examined the effects of a 1-year cryopreservation. This duration of cryopreservation is also commonly required in clinical settings. To extend the limit established by our study, the impact of longer cryopreservation periods on allografts will be examined.
Cryopreservation has been widely adopted and developed for clinical use, particularly in the field of reproductive medicine. No cells, tissues, or biological materials are able to sustain chemical reactions at absolute zero during deep cryopreservation. Cryopreservation has been applied to the preservation of cornea, blood vessels, cartilage, cardiac valves, skin, islet cells,and ovaries. Cryopreservation of nerve grafts may become a useful adjunct to clinical nerve allografting by permitting elective scheduling of surgery and more time for preoperative tissue testing (Evans et al., 1998). However, studies examining whether extremely low temperature changes the structural and biological properties of nerve grafts are scarce. Hundepool et al. (2017) stored Sprague-Dawley rat acellular nerve at either 4 ° C or −80 ° C. The results showed storage at −80 ° C heavily damaged the nerve ultrastructure compared with 4°C storage (Hundepool et al., 2017).
Currently, two widely used slow-cooling methods are employed for the cryopreservation of biological tissues, including use of an automated freezer and the “two-step” method,both of which can cool down tissue at room temperature to a temperature typical of liquid nitrogen. The “two-step”method, which was used in our study, achieves cryopreservation through a sequential placement of the tissue inside a series of refrigerators whereby the tissue is cooled by a temperature gradient. Compared with use of an automated freezer, which requires a complex operating procedure, the “two-step”method is more effective and economical and can thus be more widely adopted. After storage at extremely low temperatures, cells and tissues were brought from room temperature to the designated temperature, and then thawed to room temperature. Their intracellular and extracellular fluid would undergo phase transition, which might cause tremendous changes of osmotic pressure within cells, thus leading to physical and biochemical changes . Cells and tissues should endure these changes, especially at temperatures ranging from −15 ° C to−60° C, to remain alive. Nevertheless, most of the time, these changes were fatal to cells and tissues. Many theories have been raised by scientists regarding cryo-damage. Mazur et al.(1972) proposed two factors contributing to this type of damage: intracellular ice formation and slow cooling rate-induced solution damage. Intracellular ice that forms during freezing of biological tissues and cells is one crucial cause of direct cell injury. As cells are eluted from decellularized nerve tissue after chemical extraction, and because tissue then contains only the matrix along with the epineurium and perineurium, it is unlikely that cell or tissue damage originates from intracellular ice formation. In contrast, solution damage would cause minimal damage to acellular tissue matrix. To maintain the intactness of nerve grafts, application of a cryoprotectant is required to protect materials from being destroyed. DMSO has been commonly used for cell and tissue cryopreservation, as it reduces the formation rate of ice crystals by competing with the hydrogen atoms of water, thereby reducing the solidification of water. DMSO can minimize cell damage caused by exorbitant extracellular solute concentration during the slow freezing process as well as the destruction of cell and tissue structure by the formation of ice crystals. It has been reported that if no cryoprotectant was added to the medium during cryopreservation,growth rate of epidermal cells would decrease to 0%, while application of 15% DMSO increased the growth rate to 90% (He et al., 2010). Combined with fetal bovine serum, which provides nutrition for cells, DMSO could protect cells and tissue from cyro-damage. Sucrose can transport intracellular solutes extracellularly when cooling down and intracellularly while rewarming, which can reduce osmotic injury to cells or tissues.Therefore, this study cryopreserved ANAs in a cryoprotectant supplemented with 10% fetal bovine serum, 10% DMSO, and 5% sucrose in RPMI1640 medium at −80° C for 1 year.
Our present study detected no significant differences in the structure or mechanical properties of acellular nerve tissue matrix. Moreover, a sterility test did not detect any bacterial contamination. This suggests that cryopreservation does not change the physiochemical properties of decellularized nerve allografts. After transplanting allogeneic acellular nerve grafts that had previously cryopreserved for 1 year into rats following sciatic nerve removal, we found no changes in NF-200 immunoexpression, gastrocnemius muscle histology, or myelin sheath morphology. Our results indicate that cryopreservation maintains the function of acellular nerve allografts and can be safely and effectively used for the preservation of acellular tissues.
In this study, no significant differences in NF-200 expression,myelin thickness, or target muscle area were observed when cryopreserved acellular nerve grafts were transplanted to repair sciatic nerves defect in Sprague-Dawley rats. Indeed, cryopreservation had no obvious negative effect on nerve regeneration,and no adverse events were observed in experimental rats.These results further confirm the effectiveness and biological safety of using cryoprotectant (10% fetal bovine serum, 10%DMSO, and 5% sucrose in RPMI1640 medium) when acellular nerve grafts were preserved at extremely low temperature. This study has its limitations. Notably, unlike automated freezing methods, the “two-step” method cannot assure identical cooling curves in every cryopreservation. However,although automated freezing methods have the superiority of precise control over the whole process, the requirement for specialized equipment limits broad use. Thus, each institution must choose a suitable cooling method according to their conditions and resources.
Author contributions: YYH and XLX harvested the specimen, analyzed and interpreted the experimental data. JHL, XJH and SZ were responsible for the statistical analysis. YBX, BH and JQ established the animal models. ZWZ and BH designed the study and edited the manuscript.
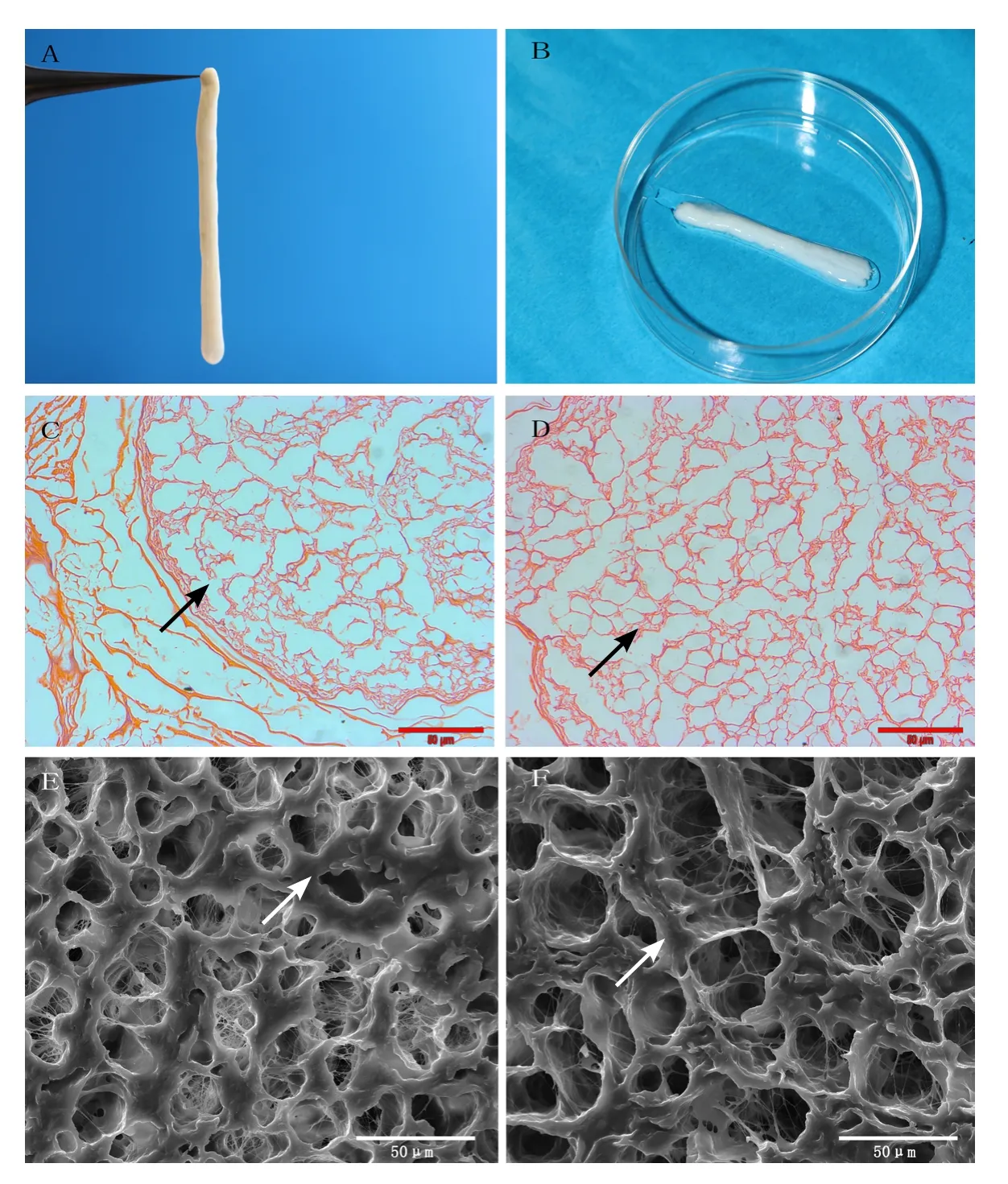
Figure 1 Observation of human acellular nerve allograft (hANA).

Figure 2 Histological analysis of rat acellular nerve allograft (rANA) on day 28 after transplantation.
QTZ, LQG and XLL revised the manuscript. All authors have read and approved the final version of the paper.
Conflicts of interest: The authors have no conflicts of interest to declare.Financial support: This study was supported by the National Natural Science Foundation of China, 81201546; the Doctoral Start-up Program of Natural Science Foundation of Guangdong Province of China,No. 2017A030310302; the Medical Scientific Research Foundation of Guangdong Province of China, No. A2016018; grants from the Science and Technology Project of Guangdong Province of China, No.2016A010103012, 2013B010404019. The funding sources had no role in study design, conection, anaysis or interpretation of data, writing and deciding to submit this paper for publication.
Institutional review board statement: The experimental protocol was approved by the Ethics Committee of the First Affiliated Hospital of Sun Yat-Sen University of China (approval number: [2013]A-055). All experimental procedures in in vivo experiments on rANA described here were in accordance with the National Institutes of Health (NIH) guidelines for the Care and Use of Laboratory Animals.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement: This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer review reports:
Reviewer 1: Michele R Colonna, Universita degli Studi di Messina, Italy.Comments to authors: This is an original report of the creation of a cryopreserved decellularized nerve allograft. The authors quote their previous studies and the present study has been performed to add novel elements to a well-established line of research. The authors base their study on sound literature about cryopreservation and the design of the study is rigorous. Statistics have been followed gaining correct numbers. The results are interesting and exciting and the Discussion shows a thorough knowledge of the topic cryopreservation.
Reviewer 2: Alonzo D Cook, Brigham Young University, USA.
Comments to authors: The article is a confirmation of the predictable outcome that cryopreservation will work for acellular nerve allografts.The dimethyl sulfoxide cryopreservation process has been shown to work for other tissues and is not novel.
杂志排行

中国神经再生研究(英文版)的其它文章
- Seeing the wood for the trees: towards improved quantification of glial cells in central nervous system tissue
- 3′-Daidzein sulfonate sodium protects against memory impairment and hippocampal damage caused by chronic cerebral hypoperfusion
- The novel chalcone analog L2H17 protects retinal ganglion cells from oxidative stress-induced apoptosis
- Treatment with NADPH oxidase inhibitor apocynin alleviates diabetic neuropathic pain in rats
- Saikosaponin a increases interleukin-10 expression and inhibits scar formation after sciatic nerve injury
- Patterns of cortical reorganization in facial synkinesis: a task functional magnetic resonance imaging study