超高效液相色谱荧光检测法测定水果中3种萘衍生物
2018-08-04仲伶俐郭灵安李华仙雷欣宇付成平
仲伶俐,李 曦,郭灵安,李华仙,雷欣宇,付成平,赵 珊
(四川省农业科学院分析测试中心,四川成都 610066)
萘衍生物1-萘乙酰胺(NAD)、1-萘乙酸(NAA)和2-萘氧乙酸(2-NOA),属生长素类植物生长调节剂,在果树上使用能加强植株的新陈代谢和光合作用,刺激生长,促进坐果和增大果粒等[1]。我国规定苹果、葡萄和荔枝中NAA最大残留限量(MRL)分别为0.1、0.1、0.05 mg/kg[2],对应的检测方法标准有SN 0346气相色谱法(GC法),检测限0.02 mg/kg[3],SN/T 2228气相色谱-质谱(GC-MS)法,检测限0.01 mg/kg,需要三甲基硅烷化重氮甲烷衍生化,操作较复杂[4]。日本规定NAA在水果、蔬菜等产品中的最大残留限量为0.03~5.0 mg/kg,NAD在苹果和梨中的限量为0.1 mg/kg[5]。欧盟规定NAD、NAA、2-NOA在浆果类水果中的最大残留限量分别为0.05、0.05、0.01 mg/kg[6]。NAA相关的测定方法已较成熟,除了上述标准方法GC法和GC-MS法外,主要有荧光法[7]、液相色谱-紫外检测法(HPLC-UV)[8-9]、液相色谱-荧光检测法(HPLC-FLD)[10]、液相色谱-串联质谱法(HPLC-MS/MS)[11-13]。而国内外对NAD和2-NOA的测定研究较少,汪伟[14]建立了NAA和2-NOA的HPLC-UV测定法。X Esparza[15]等建立了HPLC-MS/MS分析苹果中NAD、NAA和NOA的方法,并研究了施药后苹果样品中NAD、NAA和NOA残留情况及NAD和NAA的降解规律。HPLC-MS/MS灵敏度和选择性高,但仪器昂贵。水果样品基质复杂,采用HPLC-UV测定萘衍生物选择性不高,易干扰定性和定量分析。
目前我国没有NAD和2-NOA的限量规定和相应的检测方法标准,还未见有文献报道采用液相色谱-荧光检测法同时测定NAD、NAA和2-NOA。QuEChERS法作为一种快速、简便的残留提取净化技术,在农药多残留检测中得到广泛应用[16]。本文采用超高效液相色谱-荧光检测(UPLC-FLD)法,结合QuEChERS法低成本、简便快速的前处理技术,建立同时测定水果中NAD、NAA和2-NOA的方法。
1 材料与方法
1.1 材料与仪器
NAD、NAA和2-NOA标准品 纯度均>99.0%,德国Dr. Ehrenstorfer公司;甲酸 色谱纯,上海安谱实验科技股份有限公司;甲醇、乙腈、二氯甲烷 色谱纯,美国sigma-aldrich公司;氯化钠 分析纯,西陇化工有限公司;NH2固相萃取小柱 500 mg/6 mL,美国Thermo公司;PSA填料、C18填料、无水硫酸镁(MgSO4) 美国Agilent公司;实验用水 优普超纯水系统制备;草莓、苹果、柑橘、葡萄、荔枝(去核、去壳,切块后用料理机制备成匀浆状的水果样品) 市售。
Agilent 1290超高效液相色谱仪(配有荧光检测器) 美国Agilent公司;waters e2695高效液相色谱仪(配二极管阵列检测器) 美国Waters公司;LD5-2A高速离心机 北京京立离心机有限公司;WH-3微型涡旋混合仪 上海沪西分析仪器厂有限公司;DT-1002A电子天平 常熟市金羊砝码仪器有限公司;Braun Multiquick 3 K 600料理机 德国博朗公司。
1.2 实验方法
1.2.1 样品前处理
1.2.1.1 提取溶剂和提取方法的选择 提取①:称取10 g制备好的匀浆状水果样品于100 mL具塞离心管中,加入20.0 mL乙腈,涡旋2 min,加入约5 g氯化钠,盖上盖子,剧烈振摇1 min,4000 r/min离心5 min,使乙腈和水相分层。取10 mL乙腈层于50 mL梨形瓶中,于40 ℃下旋转蒸发浓缩至近干,洗耳球吹干,用4 mL二氯甲烷-乙腈(9∶1,V/V)淋洗液(A.含0.2%甲酸;B.含0.5%甲酸;C.含1%甲酸)溶解残渣,待SPE净化。
提取②:称取10 g制备好的匀浆状水果样品于100 mL具塞离心管中,加入10.0 mL乙腈溶液(A.含1%甲酸;B.含2%甲酸),涡旋2 min,加入约5 g氯化钠,盖上盖子,剧烈振摇1 min,4000 r/min离心5 min,使乙腈相和水相分层。待QuEChERS净化。
1.2.1.2 净化方法的选择 SPE净化:用5 mL二氯甲烷-乙腈(9∶1,V/V)淋洗液(A.含0.2%甲酸;B.含0.5%甲酸;C.含1%甲酸)淋洗液活化NH2固相萃取小柱,弃去流出液,活化完成后加入提取①得到的待净化样液,用50 mL梨形瓶收集流出液,再用上述淋洗液洗涤NH2固相萃取小柱2次,每次3 mL,合并流出液,于30 ℃下旋转蒸发浓缩至近干,洗耳球吹干,用乙腈-水(1∶1,V/V)溶液2 mL溶解残渣,经0.22 μm微孔滤膜过滤后,供超高效液相色谱测定。
QuEChERS净化:准确称取一定质量的吸附剂(①50 mg PSA,50 mg C18,150 mg MgSO4;②25 mg PSA,25 mg C18,150 mg MgSO4)于2 mL离心管中,移取1 mL提取②得到的上层酸性乙腈提取液于该离心管中,将提取液与吸附剂(a. 1%甲酸-乙腈,50 mg PSA,50 mg C18,150 mg MgSO4;b. 2%甲酸-乙腈,50 mg PSA,50 mg C18,150 mg MgSO4;c. 1%甲酸-乙腈,25 mg PSA,25 mg C18,150 mg MgSO4;d. 2%甲酸-乙腈,25 mg PSA,25 mg C18,150 mg MgSO4)混匀,3000 r/min离心3 min,取0.5 mL上清液,加入0.5 mL水,混匀,经0.22 μm微孔滤膜过滤后,供超高效液相色谱测定。
1.2.2 仪器条件
1.2.2.1 色谱柱的选择 比较ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×100 mm)和ACQUITY UPLC BEH phenyl(1.7 μm,2.1 mm×100 mm)2种色谱柱对化合物色谱分离的影响。
1.2.2.2 流动相的选择 比较乙腈-水和乙腈-0.1%甲酸2种流动相对色谱峰形和保留值的影响。
1.2.2.3 其它仪器条件 洗脱条件:乙腈-0.1%甲酸(25∶75,V/V)等度洗脱;流速:0.5 mL/min;荧光检测波长:Ex=280 nm,Em=330 nm;柱温:40 ℃;进样量:4 μL。
1.2.3 标准曲线的绘制 分别准确称取NAD、NAA、2-NOA标准品10.0 mg,用甲醇溶解并转移至10 mL容量瓶中,定容至刻度,配制成1.0 mg/mL的标准储备溶液,贮存于密闭的棕色玻璃瓶中。准确移取NAD、NAA和2-NOA标准储备溶液各0.5 mL于50 mL容量瓶中,用甲醇稀释成10 mg/L的混合标准溶液,并逐级稀释成0.005、0.01、0.05、0.1、0.5、1.0 mg/L的混合标准工作溶液,注入液相色谱仪,记录色谱图,以峰面积为纵坐标,以浓度为横坐标进行线性回归,绘制标准曲线。
1.2.4 加标回收实验 称取样品后加入一定量的标准溶液于样品中,使得样品中NAD、NAA和2-NOA的含量为0.01、0.1和1.0 mg/kg,混匀并静置30 min,用1.2.1方法前处理,用1.2.2方法测定。
1.3 数据处理
采用Agilent 1290Ⅱ色谱工作软件进行数据处理,得到标准曲线及回归方程,用Excel 2007绘制,色谱图由数据绘图工具OriginPro 8绘制。
2 结果与讨论
2.1 净化方法的选择
因草莓基质更加复杂,对测定的干扰相对其它水果更大,因此选取草莓样品进行净化方法的选择和优化实验。草莓样品经乙腈提取,盐析分层后进行净化操作。由于3个化合物中NAA和2-NOA都有一定的酸性,实验首先选择了具有阴离子交换作用力的NH2固相萃取小柱净化样品,当样液通过NH2小柱时,酸性化合物因离子交换作用被吸附,然后用酸性淋洗液进行洗脱,当淋洗液为含0.2%~1%甲酸的二氯甲烷-乙腈(9∶1,V/V)溶液时,NAD和NAA回收率能达到70%以上,但2-NOA回收率很低,增加洗脱溶剂的极性和洗脱体积,仍不能得到改善。后采用更加简便、快速的QuEChERS法进行前处理。样品经酸性乙腈提取,盐析分层,PSA、C18和无水硫酸镁吸附剂对提取液进行QuEChERS净化后测定。通过比较不同提取溶剂和吸附剂用量组成条件(如1.2.1中所述)下目标化合物的回收率,如图1所示,结果表明当吸附剂用量相同的条件下,提取溶剂中酸含量增加对2-NOA的回收率有明显的提高,当提取溶剂相同的条件下,减少吸附剂用量能够提高回收率。原因可能是PSA吸附剂在硅胶上键合了乙二胺丙基官能团,具有阴离子交换作用,在去除样品中的有机酸、脂肪酸和糖等干扰物的同时,对分子结构中带有羧基的化合物有一定的保留作用[17],当化合物所在溶剂环境达到一定的酸性条件下,这类化合物才能从吸附剂释放出来。最终确定以2%甲酸-乙腈为提取液,25 mg PSA、25 mg C18和150 mg MgSO4进行QuEChERS法净化,此条件下3个化合物均能获得较好的回收率。

图1 不同提取溶剂和吸附剂用量下的回收率Fig.1 The recoveries of different extraction solvents and adsorbent dosage注:a. 1%甲酸-乙腈,50 mg PSA,50 mg C18,150 mg MgSO4;b. 2%甲酸-乙腈,50 mg PSA,50 mg C18,150 mg MgSO4;c. 1%甲酸-乙腈,25 mg PSA,25 mg C18,150 mg MgSO4;d. 2%甲酸-乙腈,25 mg PSA,25 mg C18,150 mg MgSO4。
2.2 仪器条件的选择与优化
用液相色谱二极管阵列检测器扫描NAA得到其最大紫外吸收波长(UV)为220 nm,此波长选择性低。用荧光检测器对NAA进行扫描,得到其最佳激发波长(Ex)和发射波长(Em)分别为280、340 nm,用相同浓度的萘乙酸标准溶液分别在UV220 nm和Ex280 nm,Em340 nm下测定,比较萘乙酸在这两个检测器上的色谱峰响应,见图2、图3,结果表明萘乙酸在最大紫外吸收波长UV220 nm测得的色谱峰信噪比较低,荧光检测器的灵敏度明显高于紫外检测器。用荧光检测器对NAD和2-NOA进行扫描,这2个化合物的最佳激发波长和发射波长均在280 nm和330 nm附近,且NAA在发射波长为330 nm和340 nm下的响应差异很小,因此,综合3个化合物的峰响应,本方法采用选择性和灵敏度更高的荧光检测器,在Ex280 nm,Em330 nm下测定NAD、NAA和2-NOA。

图2 萘乙酸在UV220 nm的色谱图Fig.2 Chromatogram of NAA on UV220 nm
图3 萘乙酸在Ex280 nm,Em340 nm的色谱图Fig.3 Chromatogram of NAA on Ex280 nm,Em340 nm
通过比较ACQUITY UPLC BEH C18色谱柱和ACQUITY UPLC BEH phenyl色谱柱在乙腈-水,乙腈-0.1%甲酸不同流动相条件下对色谱分离的影响,发现乙腈-水为流动相时色谱峰较宽,峰形不对称,而乙腈-0.1%甲酸为流动相能够改善色谱峰形,对保留值并无影响。在相同的流动相条件下phenyl色谱柱对NAA和2-NOA的保留没有C18色谱柱强,而对NAD的保留差异不大。为了实现更快速的分离,实验选择phenyl色谱柱,以0.1%甲酸-乙腈为流动相等度洗脱,在0.5 mL/min的流速下3个化合物在6 min内能得到很好的分离,色谱峰尖锐对称(如图4所示)。由于水果品种较多,选择基质干扰较其它水果更大的草莓样品的图谱作为代表,从草莓空白样品和加标样品色谱图(图5、图6)可以看出,该条件下NAD、NAA和2-NOA在样品中基本无色谱干扰。

图4 3种萘衍生物标准色谱图Fig.4 Chromatogram of 3 naphthalene-derived compounds
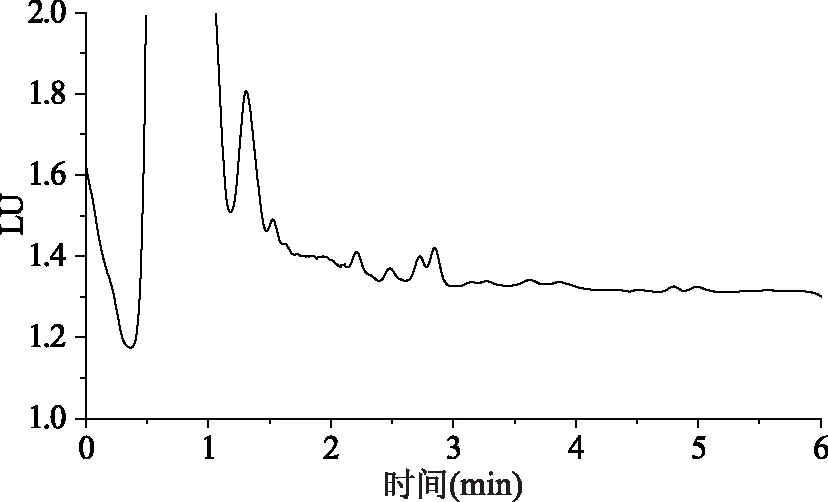
图5 草莓空白样品色谱图Fig.5 Chromatogram of the blank strawberry sample
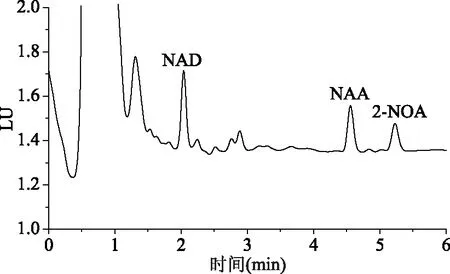
图6 草莓加标样品色谱图(0.01 mg/kg)Fig.6 Chromatogram of the spiked strawberry sample(0.01 mg/kg)
2.3 线性范围、检出限和定量限
经超高效液相色谱仪测定,3种化合物在0.005~1.0 mg/L浓度范围内与色谱峰面积呈现良好的线性关系,相关系数(r)>0.9999。通过加标回收实验,根据信噪比(S/N)≥3的峰响应值和样品前处理的稀释倍数,确定方法检出限(LOD)为0.002~0.003 mg/kg,根据信噪比(S/N)≥10的峰响应值和样品前处理的稀释倍数,确定方法定量限(LOD)为0.006~0.01 mg/kg。3种萘衍生物的回归方程、相关系数、检出限和定量限见表1。

表1 回归方程、相关系数(r)、检出限(LOD)和定量限(LOQ)Table 1 Regression equations,r,LODs and LOQs
2.4 方法的准确度与精密度
对草莓、苹果和柑橘等空白样品进行添加回收实验,添加水平为0.01、0.1和1.0 mg/kg,考察方法的准确度和精密度,回收率和相对标准偏差(RSDs)结果见表2。结果显示,3种萘衍生物在水果中的添加回收率为78.3%~95.3%,RSD为2.8%~9.5%,准确度与精密度能满足残留检测要求。
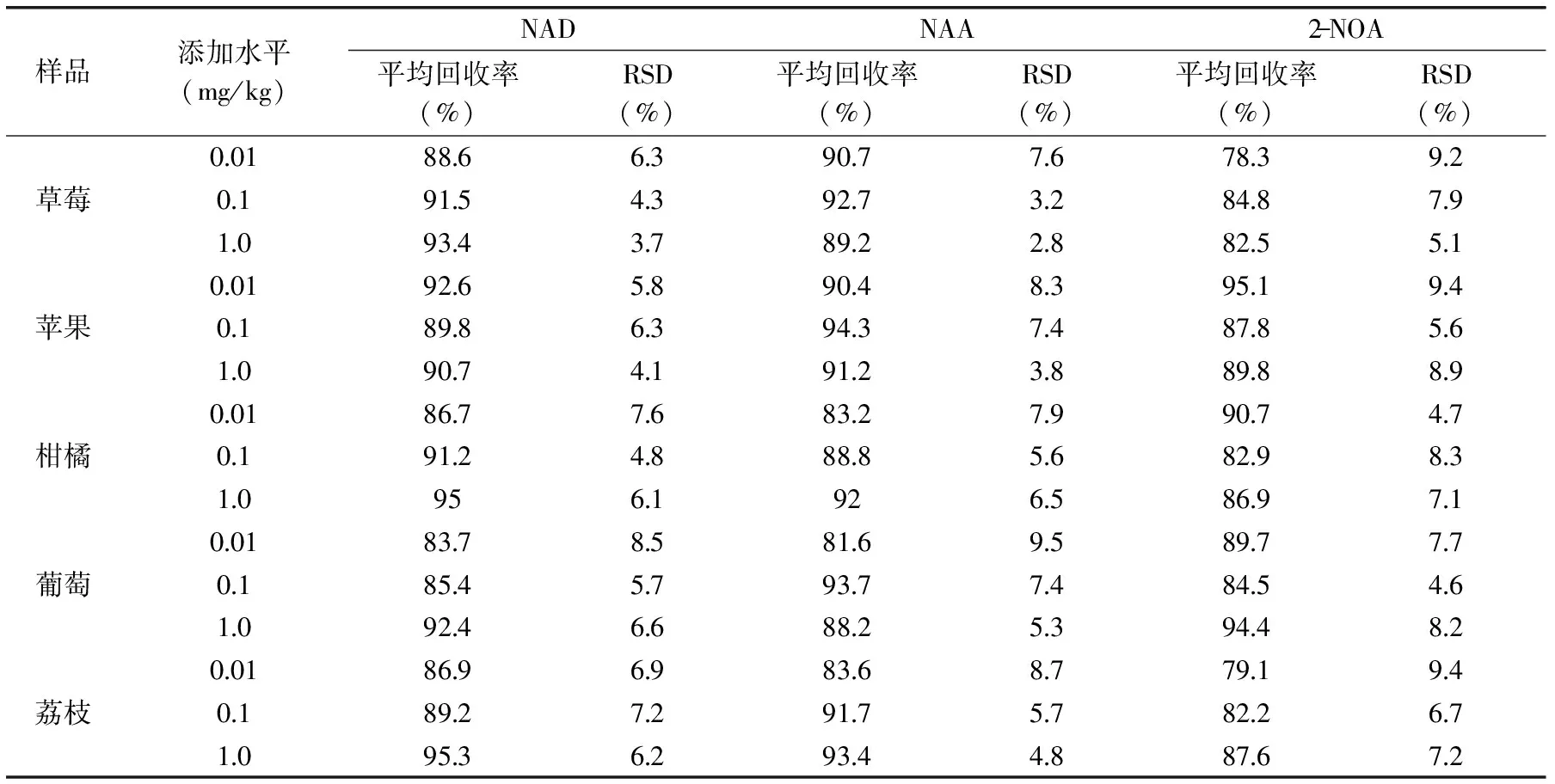
表2 添加回收率和相对标准偏差(RSD)(n=6)Table 2 Spiked recoveries and RSDs(n=6)
2.5 实际样品测定
用该方法分别对购买的草莓、苹果、柑橘、葡萄、荔枝5种水果共25个样品进行检测,结果均未检出有NAD、NAA和2-NOA残留。
3 结论
采用QuEChERS法净化,建立了超高效液相色谱-荧光法同时测定水果中1-萘乙酰胺、1-萘乙酸和2-萘氧乙酸的分析方法。该法测定水果中3种萘衍生物NAD、NAA和2-NOA,检出限可达0.002~0.003 mg/kg,比我国现有标准NAA的检测方法GC法和GC-MS法检出限更低。通过方法准确度和精密度实验得到加标回收率为78.3%~95.3%,RSD为2.8%~9.5%,准确度与精密度均符合残留分析要求,前处理操作简单、快速,选择性和灵敏度均优于HPLC-UV法,易于推广应用。
