1,8-萘酰亚胺类衍生物靶向DNA及抗肿瘤活性研究
2018-08-03胡晓琳
胡晓琳
(浙江工业大学药学院,浙江 杭州 313000)
0 引言
DNA嵌入剂是通过嵌入DNA之后,改变脱氧核糖核酸的拓扑结构,进而改变拓扑异构酶对DNA的识别功能而发挥作用,被用作抗肿瘤的研究。1980年,M.F.Brana等报导了一系列3-硝基-1,8-萘酰亚胺类化合物[1],毒理研究证明此类化合物对DNA和RNA的合成产生抑制作用,并且推断出母体嵌入DNA碱基对中,发挥抗肿瘤和抗病毒作用;从此以后,萘酰亚胺类DNA嵌入剂作为抗癌试剂的研究取得了巨大的进展。本文主要介绍为了增强1,8-萘酰亚胺与DNA的相互结合作用,以达到增强化合物在抗肿瘤方面的作用而开展的结构改造的研究进展。
1 1,8-萘酰亚胺类DNA嵌入剂
单萘酰亚胺类化合物中,最具代表性的是氨萘非特(amonafide)、米托萘胺(mitonafide),进入了治疗实体瘤的III期临床。Amonafide具有较好的阻滞P388鼠白血病以及L1210白血病肿瘤的增殖;而体外实验表明,mitonafide能有效抑制KB细胞和Hela细胞及其他一些肿瘤细胞株的生长。
1981年和1995年,M.F.Brana等分别报道了新型的1,8-萘酰亚胺类化合物[2-3],但抗肿瘤活性不及 amonafide(1)。

Mastsugo I[4]及 Staito 等[5]对 1,8-萘酰亚胺类荧光发色团做了嵌入及光敏切断的机制研究。该类化合物在366 nm光激发下能切断超螺旋DNA和寡核苷酸。
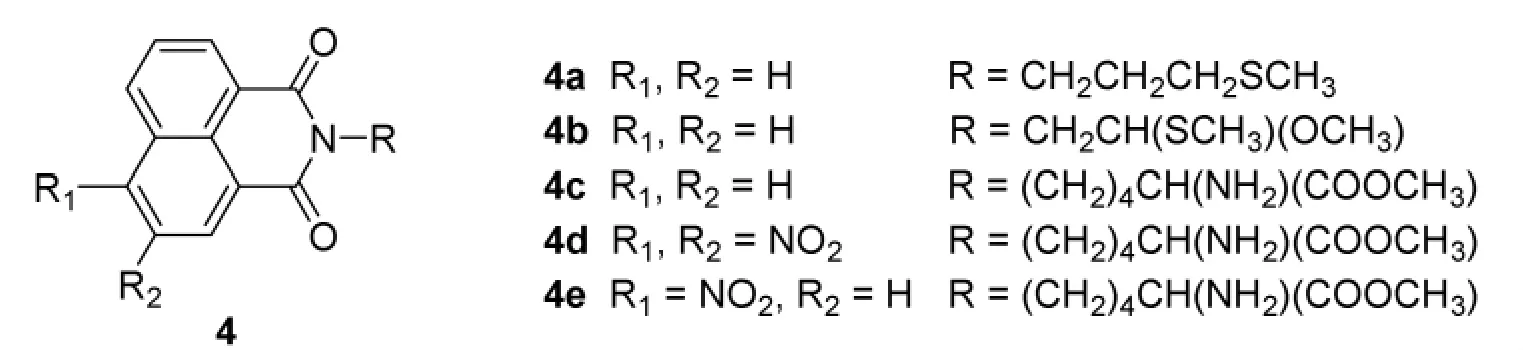
Staito等经研究得出:4d,4e能切断单链DNA和双链DNA,并且作用的位点在碱基T上的甲基,通过激发态的硝基等夺取T上甲基氢使其变为U。光照后,吡啶处理能提高其对T的切断选择性。陶志福等也对此类化合物进行了嵌入DNA的研究,通过引入呋喃环来提高嵌入性能,得到了良好的效果。
D.Wilson等用荧光淬灭技术研究了此类化合物对DNA的嵌入活性[5]得出:发色团的邻位取代基立体大小是影响嵌入常数的关键因素。取代基的体积越大,分子嵌入常数越小。

2 具有双嵌入基团的1,8-萘酰亚胺类衍生物
单萘酰亚胺衍生物Mitonafide对中枢神经系统有明显的毒副作用[6]。为了降低毒副作用,人们设计、合成了具有双萘酰亚胺类化合物6[7-9]。实验表明双-1,8-萘酰亚胺类两个母体体环双嵌入脱氧核糖核苷酸中,能专一地识别鸟嘌呤-胞嘧啶,和DNA结合力以及对肿瘤细胞的毒性效果优于单萘酰亚胺衍生物。
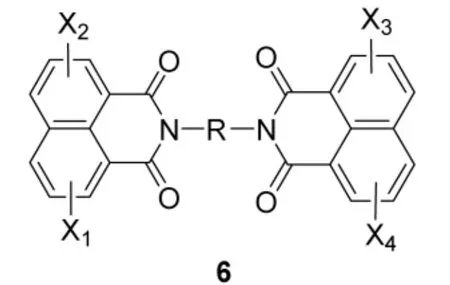
X1, X2, X3, X4为:H, NO2, NH2, C1-C6一烷氨基,C1-C6二烷氨基,OH,C1-C6烷氧基,卤素,三卤甲基,C1-C6烷基,甲酰基,C1-C6烷羰基,脲基,C1-C6-烷脲基;R为直链或支链的烷基链,链中插入一个或几个氧原子、氮原子、硫原子。
之后,化学家们对双萘酰亚胺的母体环进行了修饰,以3(或6)-硝基取代活性最好。例如,由杜邦公司开发的已进入三期临床实验的DMP840[10]。而同一位置,不同取代基也具有不同的生物活性。以3位取代基为例,活性次序:-Br>-NO2>-NH2>-H>-OH>-CONH2。近来有合成的在萘酰母环上没有任何取代基的衍生物,以LU79553对肿瘤细胞的抑效果最为突出。
由于此类化合物母体水溶性较差,不利于临床应用,通常将其制成盐的形式;适当增加linker的长度有利于母体环嵌入到DNA的碱基之间;增加链的柔性;在linker中至少有一个杂原子氮有利于增加共生物活性。
3 萘酰亚胺并环修饰
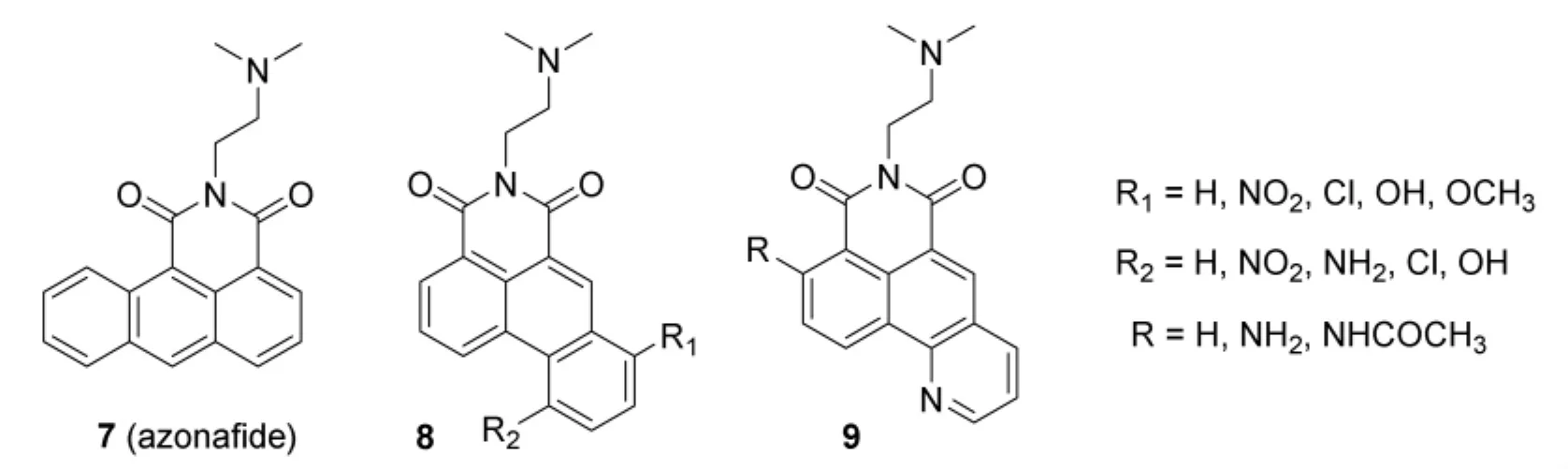
Remers研究组在阿莫萘法德结构的基础上引入了第四个环,并通过在亚胺氮上由不同的取代基取代合成19中化合物,半数化合物对小鼠和人类癌细胞的作用要强于阿莫萘法德,其中含有2-(二甲基氨基)乙基侧链的化合物azonafide(7),相较阿莫萘法德,azonafide对黑色素瘤、卵巢癌的抗增殖作用与阿莫萘法德相比,细胞毒性由10-6M数量级降低到10-7M数量级。

2003年,Brana研究组设计合成了咪唑萘酰亚胺类化合物 (10),对人结肠癌细胞HT-29的IC50为 0.94 μM,明显强于 amonafide的 4.67 μM,对HeLa和PC-3的毒性与amonafide相当。分子模拟结果显示,咪唑环堆积在DNA的胸腺嘧啶与鸟嘌呤之间,增强了化合物与DNA结合的稳定性。以缺电子的吡嗪环代替咪唑环得到的吡嗪并萘酰亚胺化合物[11](11),显示出更强的细胞毒性和与DNA的结合能力。11a对人结肠癌细胞(HT-29)的 IC50为 1.05 μM,比 amonafide 提高了5倍,对HeLa和PC-3的毒性稍强于amonafide。在萘酰亚胺环上并入呋喃、噻吩得到一系列单萘酰亚胺类化合物(12,13),这些化合物显示了较好的抗肿瘤作用,其中效果最好的化合物对人结肠癌细胞HT-29的毒性作用是elinafide的2.5倍。分子动态模拟和物理化学实验证明化合物能够与DNA形成稳定的复合物。

近十多年以来,钱旭红研究组通过在萘酰亚胺环上并入各种功能性含硫芳香杂环,取得了突出的成绩[12-13]。萘酰亚胺苯基取代咪唑类化合物14f在光切割 DNA 链,14f,15c,16a 对人肺癌细胞的毒性在10-9M数量级,显著地强于amonafide的10-3M数量级和azonafide的10-8M数量级。

作为在萘酰亚胺环上并入含硫芳香杂环研究的延续,2015年,张文研究组新合成了六个苯并[k,l]硫杂蒽-3,4-二甲酰亚胺盐酸 S1-S6[14-15],其中效果最好的S3对人肺癌A549和胃癌SGC7901 的 IC50为 0.60 μM,0.53 μM。 目前,正在进行小鼠的活体实验。
总之,尽管以萘酰亚胺为母体结构的衍生物到目前为止作为小分子药物仍然没有走上临床应用,但具有较大的成药性。
