蛋白质序列图形变换及其相似性聚类分析
2018-08-02潘以红
潘以红,钱 东,朱 平
(江南大学理学院,中国江苏无锡 214122)
人类基因组及生物信息学的发展使得DNA、RNA和蛋白质的数据库迅速扩大,寻找能够有效发现隐藏在序列数据里信息的方法变得尤为重要。蛋白质是生命的物质基础,是构成细胞的基本有机物,是生命活动的主要承担者。近年来,有关蛋白质序列图形化表示方法的研究逐渐兴起[1~4]。DNA序列由4种不同的碱基组成,而蛋白质序列由20种不同的氨基酸组成,因此蛋白质序列的图形化表示比DNA序列的图形化表示[5~10]要复杂得多。2006年,等[11]构造了一个单位“魔法”圆,将蛋白质序列转化为2D图形。他们将20种氨基酸等距离放置在圆周上,从圆心开始根据序列中氨基酸排列顺序向对应的氨基酸依次移动,类似于Jeffery[6]提出的DNA序列图形化表示法。2012年,He等[12]根据6位二进制灰度编码的顺序,引入了20种氨基酸的循环顺序,将蛋白质序列转化为3D图形,并研究了一种有效的序列相似性度量方法。2016年,He等[13]基于氨基酸的3种理化性质提出一种新的广义混沌游走表示法(chaos game representation,CGR),将蛋白质序列3D图形化。Czerniecka等[14]在前人基础上提出了蛋白质序列的20D动态图形表示法。
除了序列的图形化表示,研究者们还将其他学科的方法融入基因序列的研究。比如:动态时间归整算法(dynamic time warping,DTW)常用于孤立词语音识别,现在被应用于遗传信号的分析,Skutkova等[15,16]利用DTW对DNA信号进行分类,取得了不错的成果;离散傅里叶变换(discrete Fourier transform,DFT)常用于信号处理,近年来被广泛应用于DNA序列的分析,如基于DFT设计识别DNA序列的外显子的算法、DNA序列周期性分析等[17~20]。
氨基酸是蛋白质分子的基本组成单位,其性质决定着蛋白质的性质[21]。本文基于氨基酸的理化性质,将蛋白质序列3D图形化,经分析得到序列内氨基酸对的数量差异及序列的相似性。再将序列数值化,通过DFT等一系列算法构建出系统发育树,进而比较序列相似性。经测试,聚类结果符合实际。与其他算法相比,本文提出的序列数值化聚类方法能够保留较完整的生物信息,更加准确地反映出生物序列的相似性,便于进一步研究蛋白质序列。
1 材料与方法
1.1 数据来源
从NCBI网站的Protein Sequence下载了22条哺乳类动物的线粒体NADH脱氢酶亚基5(NADH dehydrogenase subunit 5,ND5)蛋白质序列、35条甲型流感病毒神经氨酸酶的蛋白质序列和50条冠状病毒的刺突糖蛋白序列进行研究。
1.2 方法
1.2.1 蛋白质序列图形化
蛋白质的种类很多,性质、功能各异,但都是由氨基酸按不同比例组合而成的,因此氨基酸的性质对蛋白质的性质有很大影响。本文选取氨基酸的两种主要理化性质:疏水性和相对分子质量,来构造蛋白质序列的3D模型。两种理化性质的具体数值如表1[22,23]所示。
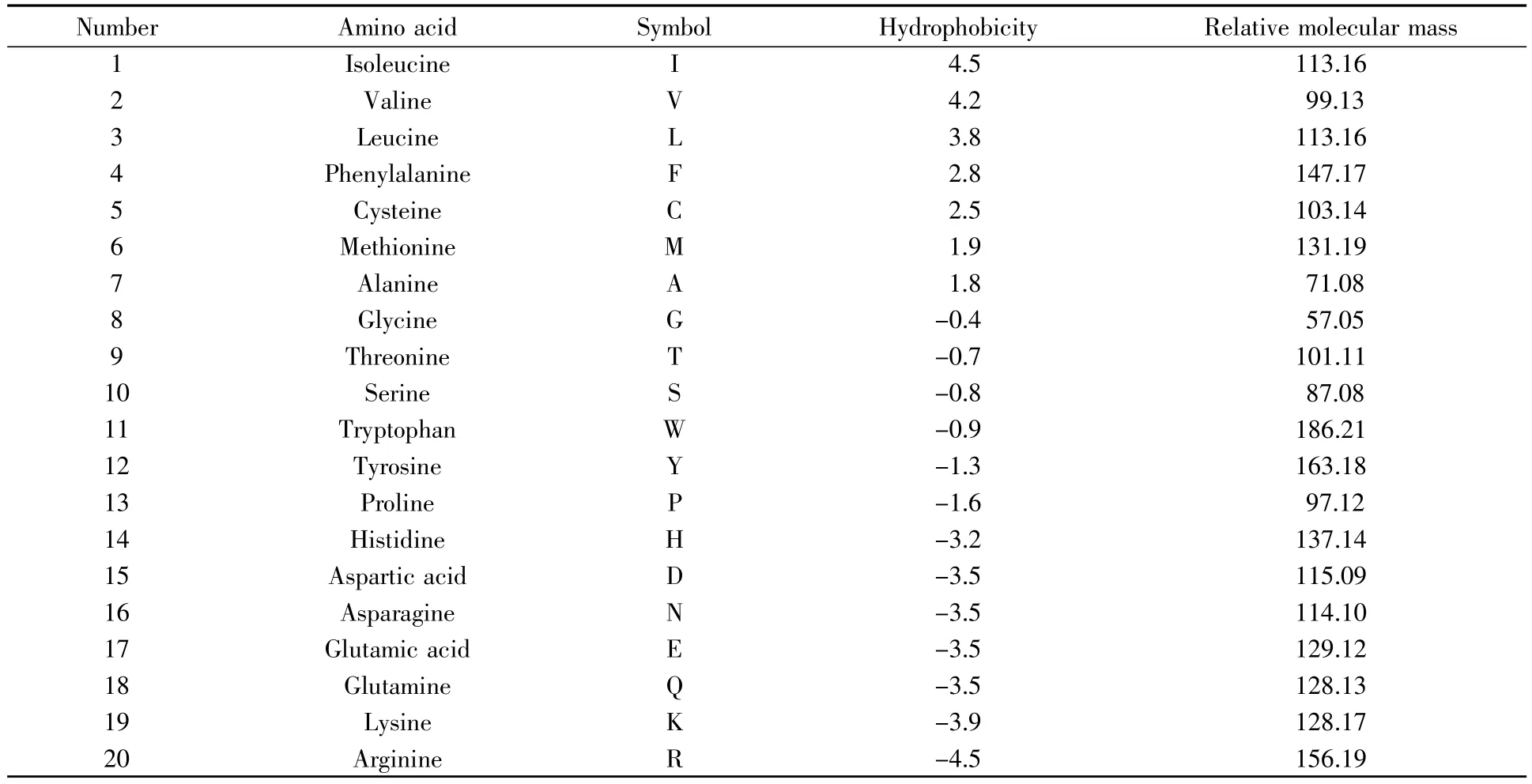
表1 20种氨基酸的疏水参数和相对分子质量[22,23]Table 1 Hydrophobicity and relative molecular mass of 20 amino acids[22,23]
本文首先将20种氨基酸根据疏水参数由大到小进行排列:I>V>L>F>C>M>A>G>T>S>W>Y>P>H>D=N=E=Q>K>R。
定义1由氨基酸的疏水参数排列顺序,可将20种氨基酸按逆时针有序地放置到单位圆周上。根据氨基酸疏水参数的整数部分大小,将氨基酸分为 8 类:{I,V}、{L}、{F,C}、{M,A}、{G,T,S,W}、{Y,P}、{H,D,N,E,Q,K}、{R}。每一类氨基酸给定一个间隔角度:间隔角度值按疏水参数大小的排列依次递减。
定义2设每种氨基酸对应圆周上的角度值为θi,θi由间隔角度累加得到,θi的取值范围是0到 2π,i=1,2,3,…,20。

图1 20种氨基酸在单位圆周上的分布Fig.1 The location of 20 amino acids on the circumfer原ence
根据氨基酸在圆周上的角度,可以得到每种氨基酸在圆周上的二维坐标:
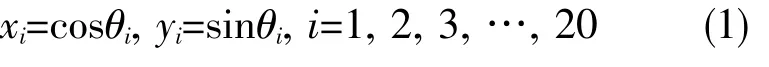
再将氨基酸根据相对分子质量排列:G<A<S<P<V<T<C<I=L<N<D<Q<K<E<M<H<F<R<Y<W。
定义3将20种氨基酸按相对分子质量的大小均分为 4 部分:{G,A,S,P,V}、{T,C,I,L,N}、{D,Q,K,E,M}、{H,F,R,Y,W},令各部分的氨基酸的z轴坐标依次
由定义2、3可得到20种氨基酸的三维空间坐标,根据蛋白质序列中氨基酸排列顺序构造出序列的3D模型。本文选取了两条不同的蛋白质序列,分别来自人类和大鼠的ND5蛋白质序列,绘制出3D模型如图2所示。通过图像比较可以发现不同序列之间存在着一些差异,比如:人类序列的3D图中氨基酸F和C之间没有连线,而大鼠序列的3D图中存在连线;人类序列的3D图中氨基酸A和P之间有连线,而大鼠序列的3D图中没有连线;两张图象中部分氨基酸之间连线密度也不一样;等等。
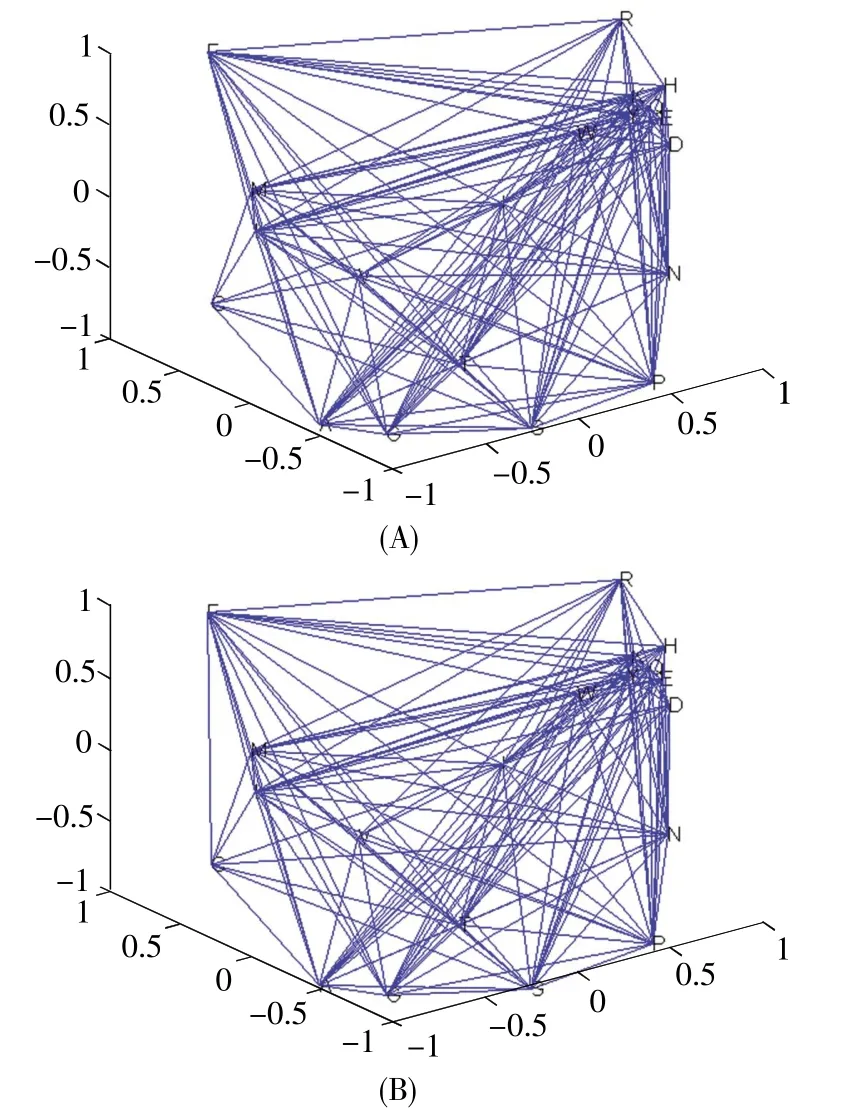
图2 人(A)和大鼠(B)ND5蛋白质序列的3D模型Fig.2 The 3D models of ND5 protein sequences of hu原man(A)and rat(B)
蛋白质的一级结构就是蛋白质多肽链中氨基酸的排列顺序,是由基因遗传密码的排列顺序所决定的。各种氨基酸按遗传密码的顺序,通过肽键连接起来。若两种氨基酸为一对,则氨基酸对有400种。由图2可以发现,不同蛋白质序列中氨基酸对数量存在差异,但对于具体数量差异特征无法清楚得知。为了进一步研究不同蛋白质序列中氨基酸对的数量差异,本文将序列的3D模型转换为20维矩阵图来表示。模型中各点之间的连线数量用颜色深浅表示(矩阵图旁标有颜色数值条)。20维即上文按疏水参数所排序的20个氨基酸:I>V>L>F>C>M>A>G>T>S>W>Y>P>H>D=N=E=Q>K>R,I对应1,R对应20。通过矩阵图来分析不同物种序列中氨基酸对的数量特征,同时初步分析序列间相似性。
1.2.2 蛋白质序列数值化及相似性比较
为了验证由分析矩阵图初步得出的序列相似度的结论,本文进一步对序列聚类方法进行研究。首先将蛋白质序列图形表示转化为数值表示。运用MATLAB 2012b软件来实现本文算法的操作。
定义4对于给定长为N的蛋白质序列S=s1s2s3…sN,根据序列中每个氨基酸的空间坐标将原序列变换为新的序列T={t1,t2,t3,…,tN},令ti=(xi,yi,zi)。序列T也可由3条子序列组成:
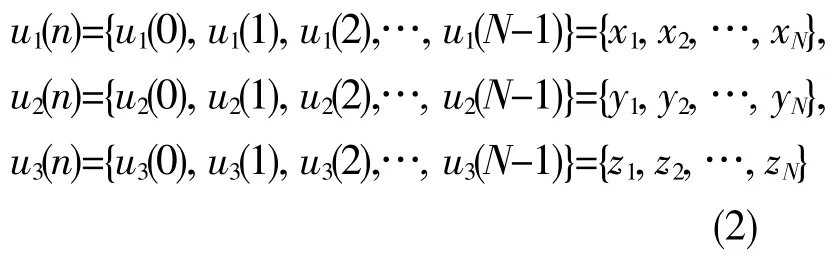
可见每条蛋白质序列都可以变换为3条数值序列,每种氨基酸都有特定的坐标,转化过程不会产生混乱,保证了信息的完整性。
DFT能够将时域信号的采样变换为频域的采样,不会丢失信号信息。通过分析序列的DFT功率谱可以发现原序列中隐藏的信息。将式(2)中的3条数值序列视作信号,分别进行DFT有:

其中,n为时域(蛋白质序列中各氨基酸位置)的离散化,k为频域的离散化。
再分别计算3条复数序列Ui(k)的平方功谱,相加得到整个蛋白质序列S的功率谱序列:
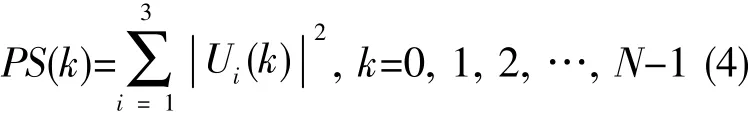
蛋白质序列不是等长度的,因而经过DFT后得到的功率谱序列也不是等长度的,不能直接进行相似性比较。Yin等[24]提出了DNA序列功率谱的扩展方法,将不同长度的功率谱序列均匀地扩展为等长的序列,具体方法如下:假设PSN是长度为N的原序列,PSM为扩展后的长度为M的新序列。则:

在Yin等[24]所研究的DNA序列数据集中,长度M是由其中最长的DNA序列决定的。这种扩展方法能够有效保留原序列的信息,也可应用于其他时间序列的扩展,但短序列的长度不能小于最长序列的一半。
本文将此方法应用于蛋白质序列功率谱的拉伸,数据集中每条蛋白质序列的功率谱长度都被均匀地扩展至数据集中最长序列的长度m。经拓展后,蛋白质序列的功率谱构成了一个新的m维空间。计算m维空间中功率谱序列间的欧氏距离来度量蛋白质序列相似性。欧氏距离d(S1,S2)越小,说明两条序列相似度越高。
蛋白质序列S1和S2的相似距离算法如下:
输入:蛋白质序列S1(长度L1),S2(长度L2),L2>L1
输出:两条序列的相似距离
算法步骤:
Step1 将序列S1、S2变换为数值序列T1和T2;
Step2 计算序列T1、T2的功率谱PS1和PS2;
Step3 将序列PS1从长度L1拓展为长度为L2的新序列PS12;
Step4 计算欧氏距离d(PS12,PS2)。
注:功率谱的零项是不参与运算的,因零项值较大,会导致计算产生误差。
最后根据欧氏距离,构建系统发育树,得到多条蛋白质序列的聚类结果。
2 结果分析
2.1 矩阵图差异分析
2.1.1 22条ND5蛋白质序列的矩阵图差异分析
本文首先选取了22条哺乳类动物ND5蛋白质序列(表2),分别做出矩阵图(图3)。
蓝鲸和鳍鲸都属于海洋哺乳类动物,且是相近的物种。观察图3-1和图3-5可以发现:两条ND5 序列中氨基酸对 L-I、L-L、L-T、T-L 数量较多,且氨基酸L含量是最多的,两者具有高度的相似性。此外,鳍鲸ND5序列中氨基酸对L-I、LT、T-T数量要明显多于蓝鲸,这表明蓝鲸和鳍鲸虽然纲目相同,但两者序列中部分氨基酸对数量存在差异。
婆罗洲猩猩、黑猩猩、长臂猿、大猩猩、人类、侏儒黑猩猩、苏门答腊猩猩都是灵长类动物,它们的矩阵图(图 3-2、3-4、3-6、3-7、3-10、3-14、3-18)也较为相似。由图可知,7条序列中氨基酸对L-L数量都是最多的。长臂猿属长臂猿科,其他都属人科,可以发现长臂猿序列中氨基酸对S-L、T-L数量明显比其他序列多。黑猩猩、侏儒黑猩猩、人类和长臂猿序列中氨基酸对L-T数量比其他序列多。因此,黑猩猩、侏儒黑猩猩和人类的序列相似度较高,长臂猿的序列与它们的相似度次之;婆罗洲猩猩、大猩猩、苏门答腊猩猩这三者相似度较高,与其他4条序列的相似度稍低。

表2 22条哺乳类动物ND5蛋白质序列信息Table 2 The information of 22 ND5 protein sequences
其他物种,比如韩国牛和西班牙牛序列中氨基酸对L-I数量最多;灰海豹和麻斑海豹、猫和老虎序列中氨基酸对L-L数量最多。矩阵图表明以上这些序列之间都具有一定的相似度。
通过对22张矩阵图的观察,发现亮点都集中在左上部分,表明ND5蛋白质序列中疏水性强的氨基酸对数量较多;同时,绝大部分序列中氨基酸对L-L的数量最多,表明ND5序列中氨基酸L含量最多;此外,所有ND5序列中疏水性最弱的氨基酸对R-R数量也相对较多。除了以上这些共性外,不同物种的序列之间还存在着差异:比如负鼠和大袋鼠矩阵图(图3-13、3-19)中亮点较多,很容易与其他矩阵图区分开,表明序列中有多种氨基酸对的数量较多;马、鸭嘴兽、犀牛的序列与其他序列相比,除了氨基酸对L-L数量最多之外,还分别含有较多氨基酸对S-L、F-L、L-T,并且由矩阵图(图 3-11、3-15、3-17)观察可知,这3条序列与其他序列相似度稍低。
2.1.2 35条甲型流感病毒蛋白质序列的矩阵图差异分析
甲型流感病毒是常见的流感病毒,容易发生变异,对人类致病性极高。本文从NCBI数据库中选取了35条甲型流感病毒蛋白质序列(表3),分别作出矩阵图(图4)。根据7种不同亚型的甲型流感病毒,选取了14条序列的矩阵图进行具体分析。
观察所选取的14张矩阵图可以发现:同种亚型的甲型流感病毒的不同序列间具有高度相似性;不同亚型的甲型流感病毒序列间存在明显差异,所含氨基酸对数量各有特点:病毒H1N1亚型序列中的氨基酸对R-R、N-G、I-G数量较多;病毒H2N2亚型序列中氨基酸对S-G、R-R数量较多;病毒H3N2亚型序列中氨基酸对S-G、S-S、R-R数量较多;病毒H5N1亚型序列中氨基酸对I-G、S-G、N-G、R-R 数量较多;病毒 H7N7亚型序列中氨基酸对T-I、G-S、N-N、R-R数量较多;病毒H7N9亚型序列中氨基酸对N-N、R-R数量较多;病毒H9N2亚型序列中氨基酸对S-I、G-T、S-G、S-S、R-R数量较多。病毒H1N1亚型和病毒H5N1亚型序列具有一定相似度;病毒H2N2亚型、病毒H3N2亚型和病毒H9N2亚型序列具有一定相似度;病毒H2N2亚型与其他亚型序列相似度稍低。
图中的亮点分布较杂乱,但所有矩阵图中心部分的亮点都较多,表明甲型流感病毒蛋白质序列中疏水性中等的氨基酸对数量较多。所有序列中疏水性最弱的氨基酸对R-R数量都相对较多,这个特征与哺乳类动物的ND5序列是相同的。
2.2 聚类结果与分析
2.2.1 22条ND5蛋白质序列聚类
使用本文的算法对2.1.1中提到的22条哺乳类动物ND5蛋白质序列进行聚类,得到了22条序列的系统发育树(图5)。Hou等[25]使用DTW方法得到的22条ND5蛋白质序列聚类结果中将老鼠和负鼠分在了一类。而负鼠是有袋类动物,应该和大袋鼠分在一类,本文的分类结果是正确的。除此之外,两种方法中的其他物种分类结果都相同,说明本文聚类算法是有效的。
本文2.1.1中由分析矩阵图初步总结的不同物种间相似性的结果与算法聚类结果是一致的,且算法所得出的聚类结果更为精细。22条序列最终被分为四类:(18,6,7,10,14,4,2,11,22,21,20,3,5,1,9,8),(16,12),(13,19,15),(17)。其中,第一大类都是大型哺乳类动物;第二类是鼠类;第三类是原始低等哺乳动物;第四类为犀科,属濒危物种。在第一大类中,又可继续细分:(18,6,7,10,14,4,2)是灵长类;(11)是奇蹄类动物;(22,21)是偶蹄类动物;(20,3)是猫科动物;(5,1)是海洋哺乳动物;(8,9)是可以水陆两栖的海洋哺乳类动物。因此,聚类结果较为精确且符合实际。
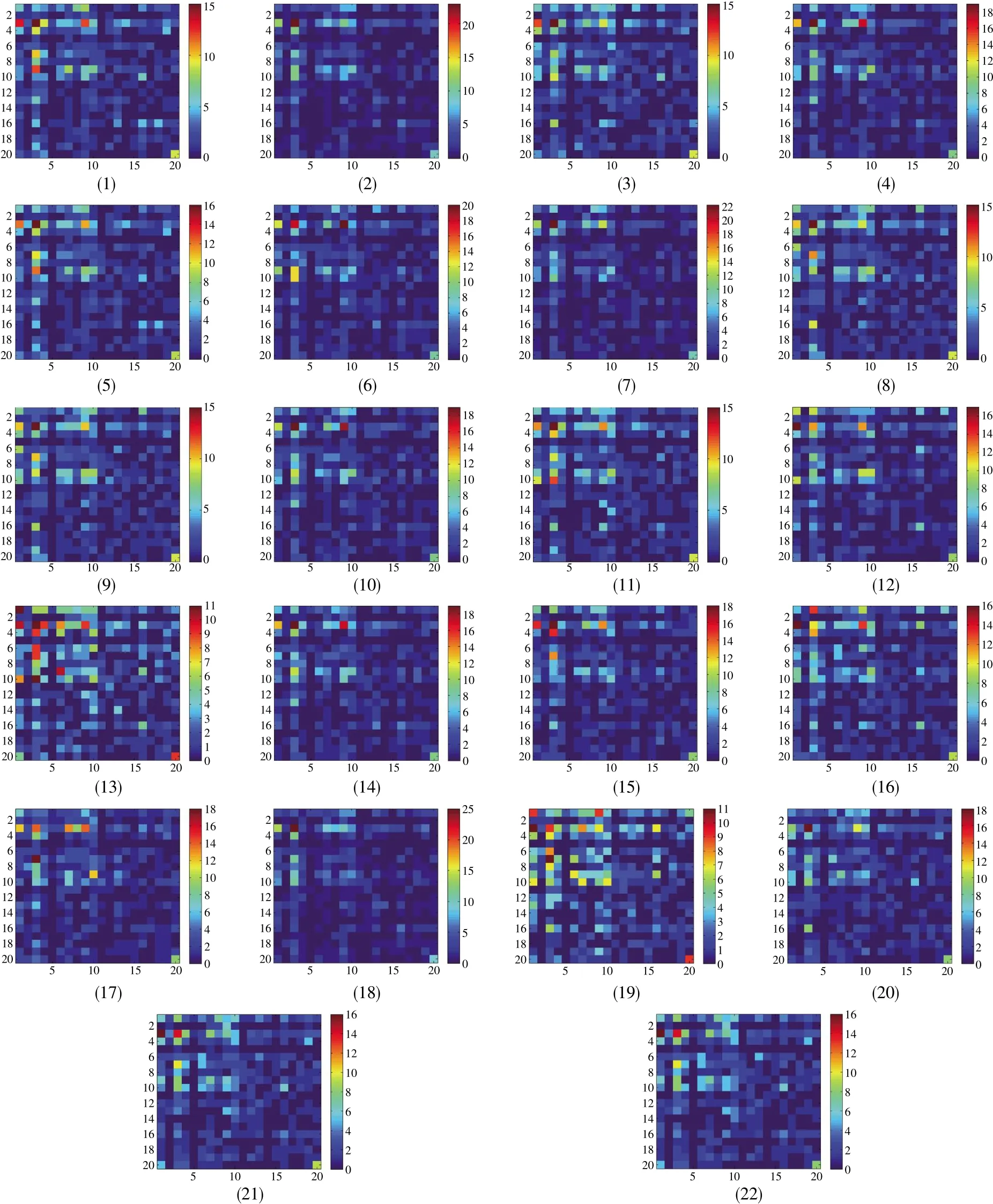
图3 22条哺乳类动物ND5蛋白质序列矩阵图Fig.3 The matrix diagrams of 22 ND5 protein sequences
2.2.2 35条甲型流感病毒蛋白质序列聚类
为了说明本文聚类算法对其他生物序列的有效性,对2.1.2中提到的35条甲型流感病毒蛋白质序列进行了聚类。图6A、B是分别用本文算法和MEGA 6.06软件构建的35条甲型流感病毒蛋白质序列的系统发育树。通过比较发现两种聚类结果具有高度相似性,大部分蛋白质序列都被准确分类,且与2.1.2中的矩阵图分析得出的相似性结果是相同的。此外,本文算法构建的系统发育树可以显示更清晰的层次结构和不同病毒之间的关系,例如:病毒序列(A/Duck/Ohio/118C/93(H1N1))属于甲型H1N1病毒,在图6B中被分类错误,但它在本文算法构建的发育树中分类是正确的(图6A)。邻近年份里出现的病毒在本文算法构建的发育树中距离更相近,例如:序列(A/turkey/O-hio/313053/2004(H3N2))与(A/Dunedin/1/2000(H3N2))的距离比(A/Washington/01/2007(H3N2))与(A/Dunedin/1/2000(H3N2))的距离更近。因此,本文的方法改进了MEGA 6.06软件分类的不足,更加具有准确性。

表3 35条甲流病毒蛋白质序列信息Table 3 The information of 35 protein sequences of influenza A viruses
2.2.3 50条冠状病毒的刺突糖蛋白序列聚类
冠状病毒会引起人类呼吸系统感染,是成人普通感冒的主要病原之一。为了进一步说明本文聚类算法的适用性,我们对50条冠状病毒的刺突糖蛋白序列进行聚类分析,数据来源于文献[26]的表3。50条序列的系统发育树如图7所示,聚类结果与文献中的结果[26]一致。所有SARS冠状病毒都聚集在同一分支,同一物种的冠状病毒的相似性更高。因此,本文的算法具有一定合理性。
以上研究表明,本文的序列数值化聚类方法蕴含了较完整的生物信息,能够较准确地反映生物序列中的相似性信息。
3 结束语
本文基于氨基酸的两种主要理化性质(疏水性和相对分子质量)构建序列的3D模型。将3D模型转换为20维矩阵图进行序列中氨基酸对数量特征及相似性分析,发现哺乳类动物线粒体ND5蛋白质序列内疏水性强的氨基酸对数量较多,甲型流感病毒蛋白质序列内疏水性中等的氨基酸对数量较多。为进一步研究不同序列的相似性,将原蛋白质序列的空间坐标转换为数值序列。对数值序列进行离散傅里叶变换,得到原序列的功率谱。再将功率谱序列均匀扩展为等长序列,根据功率谱序列间的欧氏距离来构建序列的系统发育树。最后分别对3个不同的数据集进行聚类分析,结果与实际相符。同时,将聚类结果与其他算法进行对比,证明了本文提出的聚类算法的有效性和准确性。综上所述,本文基于矩阵图的分析能够发现蛋白质序列的一些生物信息,提出的序列相似性聚类算法具有一定的可靠性。

图4 35条甲型流感病毒蛋白质序列矩阵图Fig.4 The matrix diagrams of 35 influenza A virus proteins
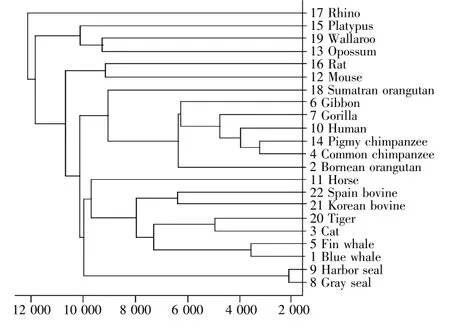
图5 22条哺乳类动物ND5蛋白质序列的系统发育树Fig.5 The phylogenetic tree of 22 ND5 protein sequences based on our algorithm

图7 50条冠状病毒的刺突糖蛋白序列的系统发育树Fig.7 The phylogenetic tree of 50 coronavirus spike proteins
