急性肾损伤,多发血管平滑肌脂肪瘤,双肺囊性病变
2018-07-27陈茹萱刘昕超徐凯峰张玉石范思远李明喜陈丽萌
陈茹萱,张 磊,刘昕超,徐凯峰,张玉石,张 夏,范思远,张 舒,李明喜,陈丽萌,郑 可
中国医学科学院 北京协和医学院 北京协和医院 1内科 2肾内科 3感染内科 4呼吸内科5泌尿外科 6眼科 7神经科 8皮肤科, 北京 100730
1 病例简介
患者女性,20岁,主诉“咳嗽18 d,发热、血肌酐升高13 d”,于2016年12月30日收住北京协和医院肾内科。
1.1 现病史
患者2016年12月12日无明确诱因出现咳嗽、咳少量黄黏痰,咳嗽时伴胸痛,无发热、憋气、咯血,当地医院考虑“肺炎”,予阿奇霉素(剂量不详)静脉输注3 d,咳嗽、咳痰稍好转。5 d后出现发热,体温最高40 ℃,无畏寒、寒战,伴头晕、乏力、多汗,咳嗽、咳痰同前,无皮疹,无自觉腹部包块、腹痛、呕吐、腹泻,小便约1500 ml/d,无尿频、尿急、尿痛,无夜尿增多及下肢水肿,自服尼美舒利退热(0.5 g,2~3次/d)。于当地医院住院治疗,测血压 110/60 mm Hg(1 mm Hg=0.133 kPa);血常规:白细胞25.08×109/L,中性粒细胞百分比88.2%,血红蛋白75 g/L,血小板336×109/L;血涂片:中性粒细胞胞浆颗粒增多增粗、可见空泡,成熟红细胞大小不等、色素充盈欠佳;尿常规:潜血(++),蛋白(-);血生化:血肌酐241 μmol/L(12月18日)→230 μmol/L(12月19日)→214 μmol/L(12月21日),血尿素氮12.49~15.19 mmol/L,乳酸脱氢酶1253 U/L,血钾、白蛋白、谷丙转氨酶均正常;降钙素原14.5 μg/L;红细胞沉降率139 mm/h,超敏C反应蛋白176.8 mg/L,铁蛋白614 ng/ml;抗中性粒细胞胞浆抗体、抗肾小球基底膜抗体(-);胸腹部CT平扫示左肺上叶少许渗出影,双肺多发小结节和小气囊,考虑炎性结节可能性大,双肾多发错构瘤,子宫发育不良,骶骨局部骨质硬化。予头孢唑肟和左氧氟沙星(剂量不详)抗感染治疗10 d,症状无改善,遂至我院就诊。发病以来,患者精神、体力、食欲差,食量减为平素的1/3,二便如常,体重无明显改变。
1.2 既往史
患者2岁时曾有2次发作性目光呆滞,对外界刺激无反应,持续数秒后自行缓解。2岁出现面部颧骨区皮肤多发棕色小结节样突起,随年龄增长逐渐增多。2015年运动中突发左侧气胸,测血红蛋白80~90 g/L,未予重视。
1.3 个人史及家族史
顺产,生长发育与同龄人相近,性格偏内向,在校大学生。不喜进食蔬菜。否认疫区旅居史,否认特殊药物及毒物接触史。父母及姐姐身体健康,家族中无类似疾病。
1.4 入院查体
体温38.5 ℃,呼吸20次/min,脉搏115次/min,血压108/65 mm Hg,血氧饱和度98%;体质量指数19.4 kg/m2;颧骨区皮肤可见多发棕色血管纤维瘤,额头皮肤可见棕色纤维斑块,上臂、胸前及腹部皮肤可见散在色素脱失斑;右手中指及双足脚趾多发甲周纤维瘤(图1);全身浅表淋巴结未触及肿大;睑结膜苍白;双肺、心脏查体无特殊;腹部稍膨隆,腹壁张力较高,双侧侧腹部可触及直径约15 cm包块,质稍硬、无压痛,肝脾肋下未及,双肾区无叩痛;双下肢无水肿;四肢肌力V级,双侧巴宾斯基征(-),脑膜刺激征(-)。
1.5 实验室检查
血常规:白细胞13.19×109/L,中性粒细胞百分比85.3%,血红蛋白65 g/L,血小板257×109/L,红细胞平均体积95 fL,红细胞平均血红蛋白浓度304 g/L。
尿常规+沉渣检查:尿蛋白微量,沉渣结果正常。
便常规+潜血:(-)。
血生化:血钾5.8 mmol/L,血肌酐208 μmol/L,血尿素氮11.9 mmol/L,白蛋白29 g/L,谷丙转氨酶88 U/L,谷草转氨酶42 U/L,乳酸脱氢酶702 U/L;血清钙2.02 mmol/L,血清磷1.79 mmol/L,血清铁28.2 μmol/L,总铁结合力214 μg/dl,转铁蛋白饱和度11.8%,血清铁蛋白288 μg/L;血清叶酸、维生素B12均正常;心肌酶(-);脑钠肽128 ng/L。
免疫指标: 红细胞沉降率>140 mm/h,超敏C反应蛋白44.97 mg/L,补体、免疫球蛋白3项、抗核抗体、血免疫固定电泳正常。

图 1 患者右手中指甲周纤维瘤(A,箭头),双足脚趾多发甲周纤维瘤(B,箭头)
感染指标:肺炎支原体抗体、痰培养(-)。
泌尿系检查:24 h尿蛋白定量650 mg;尿蛋白电泳示肾小球来源60.1%。
1.6 影像学检查
超声:双肾区正常位置未探及正常肾脏,肾盂、输尿管、膀胱未见明显异常;肾动脉彩色多普勒超声示左肾动脉近心段收缩期峰值流速高于右侧(右侧116 cm/s,左侧159 cm/s)。
腹部及盆腔CT平扫:双肾多发错构瘤(图2);双肾旋转不良;骶骨左侧及双侧髂骨多发片状高密度影。
胸部高分辨CT扫描:双肺多发薄壁囊泡影(图3),双肺多发小结节,左肺上叶少许索条影。
头颅磁共振成像:双侧大脑半球多发皮层异常信号,双侧室管膜下多发微小结节(图4),左侧尾状核头可疑钙化灶。脑电图轻度不正常,未见痫样放电。
超声心动图示少量心包积液,左室射血分数57%。心电图示窦性心动过速。
1.7 基因检测
患者存在TSC2基因第10外显子c.921delC突变,鉴于患者父母未检测到该突变,考虑患者可能为新发突变。
1.8 眼科会诊
眼底镜检查可见双眼视网膜结节(图5),光学断层相干扫描(Optical coherence tomography, OCT)提示为视网膜错构瘤。
2 多学科讨论
2.1 肾内科
患者主要表现为多系统受累。肾脏病变为双肾多发血管平滑肌脂肪瘤(angiomyolipoma,AML)、肾功能异常、蛋白尿;肾外表现包括皮肤病变为色素脱失斑、面部血管纤维瘤、指(趾)甲纤维瘤,肺部病变为双肺多发囊泡影,神经系统病变为室管膜下结节、可疑失神发作,眼部存在双侧视网膜结节,以上表现符合结节性硬化症(tuberous sclerosis complex,TSC)的诊断[1]。
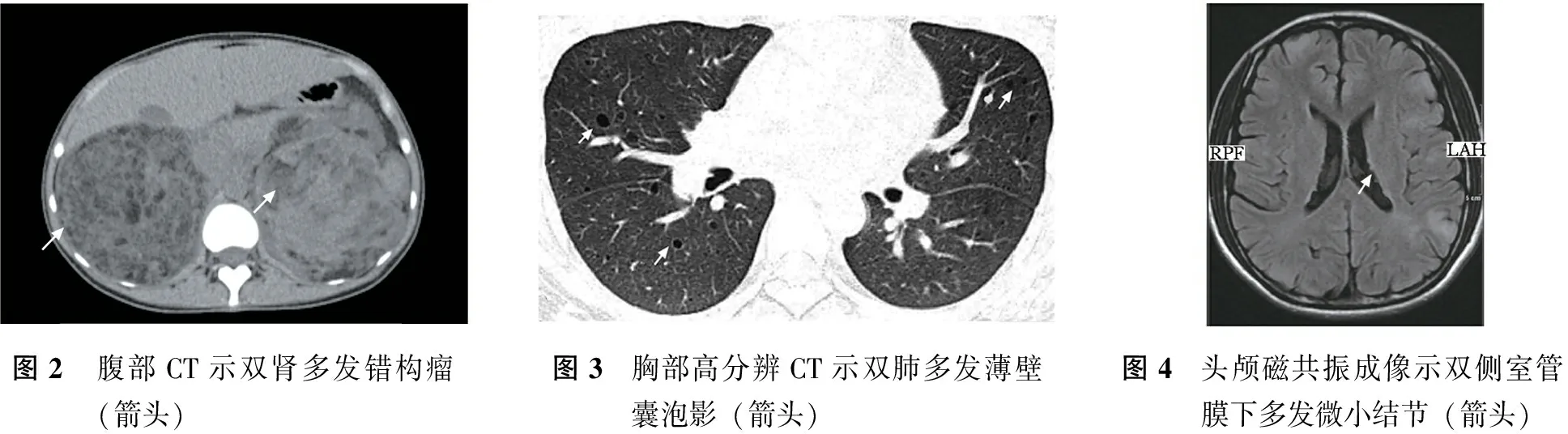
图2 腹部CT示双肾多发错构瘤(箭头)图3 胸部高分辨CT示双肺多发薄壁囊泡影(箭头)图4 头颅磁共振成像示双侧室管膜下多发微小结节(箭头)
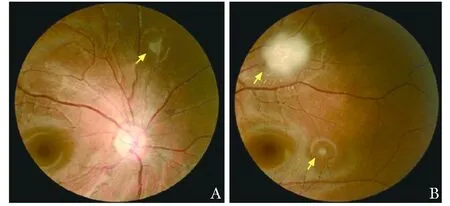
图 5 眼底检查可见双侧视网膜结节(箭头)A.右侧;B.左侧
诊断TSC需同时满足2个主要标准或1个主要标准+2个及以上次要标准。主要标准包括:(1)色素脱失斑(≥3个,直径≥5 mm);(2)面部血管纤维瘤(≥3个)或头部纤维斑块;(3)指(趾)甲纤维瘤(≥2个);(4)鲨革样斑;(5)多个视网膜错构瘤;(6)皮质发育不良;(7)室管膜下结节;(8)室管膜下巨细胞星形胶质细胞瘤;(9)心脏横纹肌瘤;(10)淋巴管平滑肌瘤(lymphangioleiomyomatosis, LAM);(11)≥2个AML。次要标准包括:(1)“斑驳样”皮肤病变(1~2 mm色素脱失斑);(2)牙釉质凹陷(>3处);(3)口内纤维瘤(≥2个);(4)视网膜无色性斑块;(5)多发肾囊肿;(6)非肾性错构瘤。患者符合7条主要标准,可诊断TSC。TSC为常染色体显性遗传病,由TSC1/TSC2基因突变导致,亦有散发病例,约75%~90%患者可检测到TSC1/TSC2基因突变[1]。TSC1/TSC2基因突变可引起哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)通路异常激活,导致细胞生长和增殖失控,形成包含血管、平滑肌及脂肪组织的肾错构瘤。本例患者行基因检测后证实存在TSC2基因突变。此外,患者起病时血肌酐241 μmol/L,1周后逐渐降至179 μmol/L,考虑起病时存在慢性肾功能不全急性加重。支持慢性肾功能不全的证据包括存在肾脏组织结构异常的慢性肾脏病(chronic kidney disease, CKD)基础及慢性贫血,该患者完善贫血相关检查后考虑为肾性贫血。
由于TSC为罕见病,我科目前针对TSC患者的治疗经验不足,希望通过此次多学科讨论制定出下一步治疗方案。
2.2 感染内科
患者发热伴咳嗽、咳痰,急性起病,结合白细胞明显升高、肺部斑片索条影,考虑存在社区获得性肺炎。经验性使用莫西沙星0.4 g/次×1次/d静脉滴注,治疗有效,体温、咳嗽、咳痰均有好转,白细胞降至正常,可足疗程给予后停用。
2.3 呼吸内科
TSC为常染色体显性遗传病,发病年龄较早,胚胎时期即可通过超声等检查发现心脏和脑部肿瘤,出生后可出现色素减退斑等皮肤病变,6个月左右可出现癫痫样发作,常有智力发育障碍,随年龄增长,逐渐出现肾脏和肺脏病变。肺部病变是TSC成年患者主要死因之一,女性多见,表现为双肺囊状及多发结节样改变。双肺囊状改变的病理基础是LAM,成年女性TSC患者发生率高达1/3以上,需注意筛查肺部高分辨CT。
本例患者以肺部感染起病,考虑为社区获得性肺炎,诊治中发现双肺LAM,结合既往气胸史及多系统表现,TSC诊断明确。
药物治疗方面,西罗莫司、依维莫司两种mTOR抑制剂在国外均已获批用于临床治疗TSC[2]。其直接针对主要发病机制环节,对各系统病变均有一定效果,且早诊断早治疗可改善患者预后,故应予以重视。根据我科诊治经验,西罗莫司与依维莫司疗效相当,而前者价格更低,可作为首选。随访中定期复查胸部影像及肺功能,血管内皮细胞生长因子D亦可作为LAM诊断和病情监测标志物。
2.4 泌尿外科
患者临床表现及基因检测结果均符合TSC。TSC患者中约80%可检测到TSC1/TSC2基因突变,国内患者以TSC2基因突变更常见,且TSC2突变患者病变常较TSC1突变患者严重[3]。肾脏病变亦为成年TSC患者死亡主要原因之一,主要并发症为肿瘤出血,出血时可考虑栓塞治疗。
根据我科诊治经验,对于AML较局限患者,可考虑手术切除肿瘤,尽可能保留正常肾脏组织,保护肾功能,但该患者双肾多发错构瘤,已失去正常肾脏组织结构,故不考虑行手术治疗。我科曾观察存在AML的成年TSC患者13例,使用依维莫司治疗,半年后肿瘤体积平均缩小超过50%,其中前3个月肿瘤缩小效果显著,1年后所有患者肿瘤体积均缩小,同时患者皮肤、神经系统病变也有改善[4]。本例患者明确存在TSC基因突变,支持mTOR抑制剂治疗。因患者近期存在肺部感染,可待感染控制后考虑使用mTOR抑制剂,因停药后病情可能反复,故建议长期甚至终身服用。
2.5 眼科
患者眼底镜检查可见双侧视网膜结节,进一步行OCT提示为视网膜错构瘤。TSC眼部病变主要表现为视网膜错构瘤(50%患者)[4]、视网膜色素减退斑,表现为视网膜中央静脉阻塞、玻璃体种植、浆液性视网膜脱离,其中后两者为继发于视网膜错构瘤的晚期表现。由于患者一般无视力下降,眼部外观无法分辨,需行散瞳检查眼底。视网膜错构瘤又称视网膜星形细胞错构瘤,是胚胎早期组织结构异常分化,神经外胚层发育障碍形成的瘤状新生物,来源于视神经纤维层。肿瘤常生长缓慢,早期透明,晚期致密、钙化,可导致播散、视网膜脱落,影响视力。本例患者视网膜错构瘤位于视网膜后极部、无钙化,根据国内及国外分类方法均为Ⅰ型。回顾我科既往开展西罗莫司治疗TSC视网膜星形细胞错构瘤经验[5],口服西罗莫司1~2 mg/d持续用药6个月以上,已使14例患者的24个错构瘤直径平均减少13.9%。建议该患者考虑西罗莫司治疗。
2.6 神经科
TSC神经系统表现主要为高级智能及认知功能减退、癫痫发作等。该患者神经系统症状较轻,幼时可疑失神发作病史,无明确癫痫发作表现,智力发育正常,认知评估未见异常,头颅磁共振成像可见皮层结节、室管膜下结节,疑似室管膜下巨细胞星形细胞瘤,头颅CT检查有助于与钙化相鉴别。建议患者定期复查头颅磁共振成像,神经外科随诊,评估有无外科处理指征。如出现癫痫发作,可至神经科就诊。
2.7 皮肤科
查体可见右额2 cm×3 cm棕色斑块,面部密集棕色丘疹,躯干及双上肢多发色素脱失斑,右手中指、双足趾甲周多发褐紫色丘疹,考虑符合TSC,面部皮疹可行激光治疗,额部斑块如有增大可行手术切除。
3 最终诊断
TSC。
4 治疗及转归
针对CKD采用非透析一体化治疗的策略:肾性贫血方面,予口服补充铁剂及叶酸等造血原料、并皮下注射重组人促红细胞生成素3000 U(2次/周);CKD骨矿物质紊乱方面,予口服钙剂纠正低钙血症与高磷血症;酸碱紊乱方面,予口服碳酸氢钠纠正代谢性酸中毒。针对肺部感染,给予莫西沙星抗感染,肺部感染完全控制后加用西罗莫司,起始剂量1 mg/d口服,1月后根据西罗莫司血药浓度水平,调整药物剂量至2 mg/d,复测血药浓度5.4 ng/ml、达到5~10 ng/ml的治疗目标范围。出院时,白细胞4.06×109/L,中性粒细胞百分比53.5%,血红蛋白95 g/L;血肌酐166 μmol/L。
门诊随访,患者精神及体力明显改善,原有皮疹无扩大、亦无新发皮疹,无视力下降,无癫痫发作。西罗莫司及CKD一体化治疗1年后,血肌酐下降至146 μmol/L,血红蛋白117 g/L,肾功能改善后逐渐减停促红细胞生成素。复查胸腹部CT:双肺多发薄壁囊泡影无增多增大、双肾多发AML无明显变化。
5 讨论
本例患者因血肌酐升高就诊,存在双肾AML、双肺LAM、多发皮肤病变、双侧视网膜结节、颅内室管膜下结节等多系统病变,最终基因检测结果证实患者TSC2基因突变,TSC诊断明确。TSC为罕见病,临床治疗经验相对缺乏。通过多学科讨论,共同制定患者诊疗计划。
TSC最初于1862年由德国病理科医生Friedrich Daniel von Recklinghausen发现,其描述了一个婴儿同时存在心脏“肌瘤”和部分硬化的脑组织。随后,法国神经科医生Désiré-Magloire Bourneville进一步描述了相关神经系统表现及合并的皮肤表现。此后,该病被命名为TSC,又称Bourneville病[6]。TSC为遗传病,患者因TSC1/TSC2基因突变,激活下游mTOR信号通路,导致各组织内细胞生长和增殖失控,引起肿瘤形成。
本病例的诊治主要围绕肾功能异常展开。虽然多数TSC患者合并肾脏AML,但肾功能不全发生率并不高。肾功能改变首先应鉴别急慢性,即区分急性肾损伤(acute kidney injury, AKI)与CKD,常用鉴别点包括有无肾脏病病史、肾脏体积是否缩小(占位性病变、糖尿病肾病除外)、有无慢性肾脏病相关并发症表现(肾性贫血、骨矿物质代谢异常等)。患者初诊时即发现血肌酐升高,因无既往肾脏资料,出现肾功能损伤的确切病程不详,故首先应寻找AKI原因。AKI病因定位分为:肾前性、肾性、肾后性。患者本次起病时因合并呼吸道感染,出现高热、并使用非甾体类抗炎药退热、进食量减少,均是可能引起AKI的肾前性因素。结合患者尿量无减少、泌尿系超声检查未见肾后性梗阻证据,考虑无肾后性因素参与。通过增加入量、停用非甾体类抗炎药等对症支持治疗后,患者血肌酐下降,但尚未降至正常范围,从治疗反应上提示应对患者进行AKI之外的CKD基础病诊断。结合影像学提示双肾AML改变、已有慢性贫血表现,明确该患者为CKD。患者存在双侧AML,致正常肾脏组织破坏而造成CKD;同时部分TSC患者还可存在肾间质纺锤状平滑肌细胞和成纤维细胞增生致肾小管间质疾病而加重CKD。因此,TSC正是该患者CKD的原因。而其出现蛋白尿的原因与CKD原因一致,为AML导致有效肾单位减少,健存肾单位负担加重,从而出现高滤过、甚至继发性局灶节段性肾小球硬化所致。
TSC常见临床表现包括腹痛、血压升高、肾功能异常、腹膜后出血、镜下出血等[7]。累及肾脏时最常表现为AML,其次为肾囊肿、肾细胞癌,肾间质性病变、局灶节段性肾小球硬化等表现少见。由于AML可引起严重腹膜后出血或终末期肾病,目前认为,AML是引起TSC相关死亡的最主要病因[6-7]。此类患者肾脏影像学检查对发现肾脏受累与监测病情变化至关重要,高危患者应警惕AML转化为恶性,同时应定期监测肾功能与尿蛋白。
主要治疗措施包括使用mTOR抑制剂、CKD非透析治疗、肾脏替代治疗等,出血时可采用栓塞治疗。随访中应注意监测患者血压、肾功能、AML瘤体大小(因超声对于缺乏脂肪组织的AML识别能力较差,故建议采用CT或磁共振成像)。
6 专家点评
肾内科郑可主治医师TSC为罕见病,尚无国内人群发病率及患病率数据。近年来对TSC的治疗有较大进展,早诊断、早治疗可明显改善患者预后。临床工作中遇到肾脏AML、肺LAM、典型皮肤病变或神经系统病变者,尤其同时存在多系统受累患者,应注意及时识别是否存在TSC并对其作出系统评估。TSC基因检测除明确诊断外,还可辅助指导治疗(有TSC基因突变更支持mTOR抑制剂治疗)及判断预后(TSC2基因突变者病变常较重)。本例患者有典型肾脏、肺部、皮肤、神经系统影像学改变,TSC诊断明确,但治疗方面仍存在难点,通过本次MDT讨论,从多学科、多角度综合考虑,更全面地为患者制定出诊疗计划。
