SASH1与肿瘤关系的研究进展
2018-07-23胥博闻综述奎翔王燕审校
胥博闻 综述,奎翔,王燕审校
650101 昆明, 昆明医科大学附属第二医院 病理科
2003年Zeller等研究人员在研究乳腺癌细胞杂交性丢失时,发现一种新的基因,该基因位于人染色体6q24-25之间,同时,研究发现相较于正常细胞,在其研究的乳腺癌病例的癌细胞中,19例患者该基因的表达产物几乎完全缺失,另46例患者则有不同程度的表达减少趋势,提出该基因有可能是乳腺癌的新的备选抑癌基因,此后,Zeller通过分析该蛋白的结构域,将其命名为SAM-and SH3-domain containing 1[1]。近年来,研究发现SASH1基因的产物在体内多种正常组织内广泛丰富表达,而在乳腺癌[1]、甲状腺癌[2]、肝癌[3]、胃癌[4]等疾病的研究中,SASH1存在表达异常或表达缺失的现象。同时,研究提示SASH1可能通过数种信号通路参与了细胞的生理现象如增殖、凋亡、细胞分化等,因此认为SASH1可能参与多种疾病的发生发展。目前国内有作者就SASH1所涉及信号通路情况进行综述[5],国内外关于SASH1与肿瘤发病关系的综述较陈旧。本文拟结合国内外最新研究成果,探讨该备选抑癌基因在肿瘤发生发展中的作用。
1 SASH1基因定位与蛋白结构
SASH1基因位于人染色体6q24.3,邻近SAMD5基因的位置。包括了22个外显子,其转录mRNA产物包括长度为7.5kb和4.4kb大小不等的两种,其中7.5kb产物表达较少见,目前仅在脑组织中发现其存在,而4.4kb产物则在除淋巴细胞和树突细胞以外的正常组织内普遍大量表达[1]。SASH1编码蛋白分子量140kda,含有1 247个氨基酸,其结构特殊,包括两个互相靠近的SAM(sterile alpha motif)结构域,以及一个鸡肉瘤病毒同源3(Src homology 3,SH3)结构域(图1)。
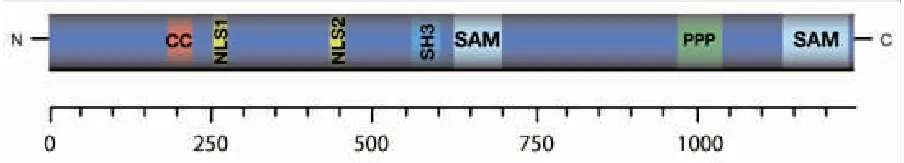
图1 SASH1蛋白结构
一些蛋白中的SAM结构域可彼此之间相互结合,形成同源二聚体,也可与其他蛋白的非SAM结构域形成特殊的非同源二聚体,通过二聚体完成信号传导的过程[6]。SH3结构域可以通过特异性结合含脯氨酸和疏水性氨基酸序列的靶蛋白,在包括RAS蛋白(rat sarcoma,Ras)、Src激酶(sarcoma gene kinases)等重要的肿瘤相关蛋白质中发挥功能,负责信号传导、调节蛋白相互作用[6-7]。因此,SASH1基因编码的蛋白产物的功用可能与这两种结构域有关,Beer等[8]认为应将SASH1纳入同样具有SH3结构域的包括SLY1、SLY2的SLY支架蛋白家族。大部分支架蛋白可与多种分子结合形成复杂复合物,以此调整细胞信号传导。研究发现SASH1蛋白可以调节细胞伪足的活动,其机制可能是SASH1的特殊结构域使其与细胞皮质区肌动蛋白相结合[9]。
2 SASH1与肿瘤
2.1 SASH1与上皮细胞间充质转化
当细胞的表型逐渐由上皮细胞转化为间质细胞时,细胞内产生的一系列连锁反应导致的现象,被称作上皮细胞间充质转化(epithelial-mesenchymal transition, EMT)。与EMT相反的过程,即细胞的间质细胞表型逐渐减弱,趋向上皮细胞变化时,则称为间充质细胞上皮化(mesenchymal-epithelial transition,MET)。EMT能够分为3型,I型能够影响胚胎的发育,II型与炎症反应有关,可以调节创伤、再生等,而III型则被认为和肿瘤的侵袭转移的病理活动存在相当重要的关系[10]。恶性肿瘤上皮细胞的顶-底极性、细胞间强粘附、运动能力偏低等高分化表型特性被逐渐消除,反而逐渐取得间充质细胞的一些重要特点,例如大量经体液移动、抗细胞凋亡、侵袭力强等。当这些细胞由于间充质特性而经过血液、淋巴液等转移至其他组织器官后,又可通过MET重新获得上皮细胞的粘附力、增殖性,继而在新的组织器官内形成上皮细胞转移灶[11]。EMT的过程复杂,涉及大量交错的信号通路过程,其机制在众多实验中被探讨,但迄今为止并不能够详细阐明,其中Heldin等[12]的研究指出,转化生长因子-β( transforming growth factor-β,TGF-β)是其重要因素之一。目前TFG-β的功能已被确认,可通过多种信号传导机制诱导EMT。其中,PI3K/AKT信号通路被TGF-β磷酸化激活是导致EMT进展的重要因素[13]。在肿瘤组织中,由于血管重塑和代谢改变,往往出现缺氧微环境,缺氧诱导因子-1α(hypoxia inducible factor-1α,HIF-1α)的大量产生可以导致包括PI3K/AKT等信号通路的磷酸化激活,进而引起肿瘤组织EMT现象[14]。EMT甚至是部分肿瘤在治疗中逐渐出现获得性耐药的重要因素之一[15]。在EMT的过程中,实验人员可以通过检测EMT的3种标志物来快捷判断EMT的进展。其中上皮来源细胞的间充质细胞标志物N-钙粘素(n-cadherin)和波形蛋白(vimentin)大量增加,而相反,上皮细胞的特色标志物E-钙粘素(E-cadherin)减少,这是EMT的最具代表性的检测手段[16]。
Zong等[4]通过实验验证,在人胃癌细胞株中SASH1基因表达降低,并且发现TGF-β能够抑制SASH1基因表达;当使用载体DNA引入大量SASH1后,TGF-β表达则受到抑制,并且下游的PI3K/AKT信号通路磷酸化物p-PI3K/p-AKT也相应减少,最终发现SASH1过表达细胞株中E-钙粘素上升,N-钙粘素与波形蛋白降低,由此推断SASH1基因过表达可以抑制胃癌细胞株发生EMT的程度。Sun等[2]在甲状腺癌细胞株的培养中观察到SASH1基因在甲状腺癌细胞中同样能够通过降低PI3K/AKT的磷酸化产物来减少EMT的发生。有研究[17]观察到在缺氧环境下的胰腺癌细胞中,SASH1有效地抑制了HIF-1α的生成量,使细胞株中EMT指标较对照组明显降低。而在一项肝癌的研究[18]中,研究人员发现,SASH1可能对肝癌细胞中另一条信号通路既阻抑胚胎发育相关信号通路(Sonic hedgehog,Shh)进行抑制,使Shh、Smo(Smoothened,Smo)等Shh信号通路关键蛋白的表达明显下降。Shh信号通路是脊椎动物hedgehog信号通路家族成员之一,主要由分泌型糖蛋白Shh配体、Smo以及下游转录因子蛋白组成。同时,已有研究[19]证实Shh信号通路异常激发伴随着肝癌细胞中由缺氧诱导的EMT,当使用抑制剂抑制Smo蛋白后,肝癌细胞EMT现象也随之受到抑制。而进一步研究[20]中,发现肝癌细胞中SASH1可能同时通过Shh信号通路和PI3K/AKT信号通路来调控肝癌细胞的EMT过程。由此推测,SASH1在肿瘤细胞进展中,具有抑制EMT发生的作用,进而可能使肿瘤的侵袭转移能力受到一定程度的限制,继而发挥抑癌效果。EMT的现象之所以在一部分恶性肿瘤的癌细胞中不受控制,也许是由于SASH1的突变所导致。
2.2 SASH1与细胞凋亡
细胞凋亡(apoptpsis)是正常细胞为了让机体能够适应环境而使老化的细胞选择主动死亡的行为。通过细胞凋亡,机体能够清除老化细胞,清除与机体不适应的细胞,甚至清除有潜在危险的细胞,如具有癌化可能的细胞。自Kerr等[21]学者在1972年首次提出细胞凋亡这一概念后,已有大量研究证实在肿瘤的进展中,肿瘤体现出的永生性表现与肿瘤细胞的凋亡异常密切相关。细胞凋亡的信号通路已知涉及包括线粒体通路[22]、死亡受体通路[23]、内质网通路[24]在内等多种通路。近年来,研究认为,基质金属蛋白酶-2和基质金属蛋白酶-9(matrixmetalloprotein-2/9,MMP-2/9)在多种肿瘤中具有使肿瘤细胞增殖活跃抗凋亡的作用,甚至参与EMT的发展[25]。对PI3K/AKT经典信号通路进行研究时,研究人员发现,AKT能够通过磷酸化细胞凋亡效应蛋白的重要一员caspase-9蛋白的Ser196位点,使caspase-9蛋白失活,从而达到抑制细胞凋亡的作用,也可以有效阻断BCL-2家族成员BAD蛋白的促凋亡作用,提示PI3K/AKT信号通路大量激活可能引起细胞失去正常凋亡能力[26],而局部粘附斑激酶(focal adhesion kinase,FAK)信号通路也可以通过干预PI3K来调控细胞凋亡[27]。作为涉及细胞凋亡的大部分信号通路的反应蛋白,细胞内Caspases家族蛋白激活,使细胞内重要蛋白水解,被认为是多条通路对细胞凋亡进行调节的共同最终途径。
Chen等[28]在对肺癌的研究中发现,A549肺癌细胞株中SASH1表达水平是降低的,引入过表达SASH1基因的A549肺癌细胞相比对照组凋亡率明显增加,而进一步干扰SASH1表达后,肿瘤细胞凋亡明显减少而增殖活跃,提示SASH1的表达参与调节细胞凋亡,他们因此提出SASH1可能是肿瘤抗凋亡能力的一种调控因素,认为涉及SASH1的信号通路具有研究的价值。另有研究发现在神经胶质瘤细胞株中,SASH1过表达的细胞株中MMP-2/9明显降低,而Caspases-3则显著上升,由此推断SASH1可能可以干预神经胶质瘤细胞的增殖、凋亡[29]。此外,在骨肉瘤[30]、黑色素瘤[31]、卵巢癌[32]细胞的体外培养中均发现当SASH1过表达后,肿瘤细胞的凋亡率出现了一定程度增加的趋势。Chen等[33]的研究中发现,SASH1可以通过抑制局部FAK信号通路,对宫颈癌细胞的肿瘤细胞学行为如增殖、侵袭等进行调控,进而抑制宫颈癌的进一步发展,提示SASH1的促肿瘤细胞凋亡能力可能与FAK信号通路有关。Burgess等[34]针对乳腺癌中SASH1的功能做进一步研究时发现,SASH1沉默的乳腺癌细胞株中氯吡拉敏抗肿瘤效果不佳,而当单独对肿瘤细胞株应用氯吡拉敏后,肿瘤细胞SASH1表达上调同时细胞凋亡率升高。已有前期研究指出氯吡拉敏能够通过FAK信号通路发挥抗肿瘤效果[35]。由此提示氯吡拉敏可能依赖SASH1通过FAK信号通路发挥抗肿瘤作用,SASH1可能作为乳腺癌氯吡拉敏疗效不敏感时的一个可靠备选治疗靶点。在此基础上进一步研究SASH1与肿瘤细胞凋亡的关系[36],发现核因子(nuclear factor- κB,NF-κB)抑制剂会使SASH1过表达的肿瘤细胞重新获得抗凋亡能力,因此判断SASH1促进肿瘤细胞调亡的能力可能与NF-κB相关信号通路相关。由此可见,SASH1基因在肿瘤细胞中表达受到抑制这一基因事件,可能是恶性肿瘤细胞获得抗凋亡能力、永生性的重要原因,SASH1蛋白可能是调控肿瘤凋亡的众多信号通路之间重要的桥梁蛋白。
2.3 SASH1与甲基化
SASH1基因在肿瘤中究竟是如何表达受阻,目前仅有猜测,并未能取得足够证据,数种猜测中基因甲基化(DNA methylation)被认为是可能性最大的重要原因。
基因甲基化是依赖于甲基转移酶的重要分子行为,是研究在DNA序列不变情况下仍发生可遗传变异的表观遗传学(epigenetics)的重要研究对象。在特定的内环境下,CpG二核苷酸5’端的胞嘧啶在甲基转移酶的催化下,被添加来自S-腺苷甲硫氨酸(S-adenosyl methionine)的甲基后形成5-甲基胞嘧啶,使大部分目标基因因此而沉默。人体内CpG位点数量巨大,其中大部分CpG位点处于甲基化状态,而处于去甲基化的CpG由于经常成簇出现,被称为CpG岛,多位于基因的启动子区域[37]。当抑癌基因启动子区域的CpG岛发生甲基化,可能导致抑癌基因蛋白表达下降,继而为癌变发生提供条件。目前DNA甲基化、DNA序列缺失、基因位点突变被认为是抑癌基因失活最重要的三种途径。已被证实在肿瘤的发生阶段伴随着某种或多种基因的甲基化,且甲基化的发生是一个多因素影响的结果[38]。
Sheyu等[39]研究了SASH1在乳腺癌中甲基化的情况,发现乳腺癌细胞中SASH1的表达下调伴随着其启动子区域甲基化,甲基化位点众多但其中导致SASH1蛋白表达下降的最主要位点为CpG_54.55和CpG_26.27,而甲基化转移酶抑制剂地西他滨有效地提高了细胞内SASH1转录RNA的含量,因此怀疑乳腺癌细胞中SASH1沉默的可能原因就是启动子区域CpG_54.55和CpG_26.27的甲基化。Peng等[3]在对肝癌中SASH1的启动子甲基化进行研究时,同样发现大量甲基化位点,而其中最重要的位点仍是CpG_26.27,是肝癌细胞株中SASH1表达受抑制的重要原因,并且地西他滨治疗同样有效。Roos等[40]在研究表观遗传学时,发现在罹患肿瘤情况不同的同卵双胞胎患者的血液中,可以检测到一个可能位于或临近SASH1启动子的甲基化位点,在双胞胎之间有着一定程度的差别,认为有深入研究的价值。目前研究认为SASH1表达下降的重要原因可能是启动子甲基化,并且能够被甲基转移酶抑制剂调控,这为以后SASH1表达下降的肿瘤的治疗提供了方向。
2.4 SASH1与基因融合
基因融合(gene fusion)是指两个或多个基因的部分序列由于染色体的移位,发生交错、重排、融合,形成新的杂合基因,并可出现新的融合蛋白。基因融合最早在1971年研究发现,在慢性髓性白血病患者外周血细胞中观察到的ph染色体是22号染色体长臂缺失了大段后的残余部分,1973年时,发现缺失的部分会移动到9号染色体长臂末端,该现象直到1982年的一些研究后首次确认ph染色体的实质为22号染色体的BCL基因与9号染色体末端的ABL基因断裂、相互异位连接的产物,于是提出了基因融合这一概念[41]。继BCL/ABL融合基因之后,目前已发现多种基因融合,涉及众多肿瘤亚型的基因组改变。这些基因融合的鉴定在肿瘤诊断和预后评估中扮演重要角色[42]。Sa等[43]在对颅底脊索瘤的基因结构重排和转录情况进行检测时,发现5种细胞株中,4种均观察到SASH1基因与位于6q24的SAMD5基因发生融合,这是第一次发现SASH1基因在肿瘤细胞中发生基因融合现象,因此实验人员推测SASH1与SAMD5的基因融合可能参与了颅底脊索瘤的发生、发展,但并未在临床体内环境内发现这种情况,因此尚需要进一步的实验验证。SASH1基因在其他疾病中的基因融合现象还需要更多的研究进行探索。
3 总 结
目前已经发现SASH1基因在正常细胞中发挥了一定的调控作用,而在多种恶性肿瘤中被观察到低表达。当SASH1过表达于多种肿瘤细胞株中后,发现肿瘤细胞的上皮细胞间质化受到抑制,而相反,发生上皮细胞间质化的肿瘤细胞株存在SASH1的表达缺失或表达下降,因此研究人员认为在正常细胞中大量表达的SASH1能够通过减少EMT的发生,从而使上皮细胞避免低分化转变,而肿瘤细胞的侵袭、转移能力或许与SASH1的低表达有关,但具体过程目前尚未有定论。已知SASH1可能通过TGF-β介导,对下游PI3K/AKT通路产生影响,发挥抗EMT作用,但具体作用机制并无定论,且可能并不是唯一通路,如Shh通路也是SASH1抑制EMT的备选信号通路。此外,SASH1表达缺失的实验室细胞株中,肿瘤细胞凋亡调控失控,实验人员提高细胞株内SASH1蛋白的水平可以令细胞凋亡率增加,提示SASH1过表达或可使肿瘤细胞失去永生性,目前提出SASH1在调控细胞凋亡过程中参与FAK信号通路与MMP-9信号通路,其功能可能依赖核因子,而其具体上下游的信号蛋白、是否参与其他通路、能否通过药物对此靶点进行控制仍有待研究。同时,已经发现部分乳腺癌患者对氯吡拉敏治疗不敏感,而SASH1基因突变有可能是导致这一现象的原因,目前研究正以此为根据找寻新的氯吡拉敏类似物[44]。研究认为SASH1的启动子区域发生甲基化是SASH1在肿瘤细胞中沉默的重要原因,那么SASH1的甲基化是否是直接导致肿瘤发生的原因之一,未来的研究需要进一步验证这一点。目前对SASH1的研究逐渐由体外转向体内实验,并且在有关宫颈癌的一份回顾性报告中,提出SASH1可以作为单独的预后预测因素[45]。另有研究指出SASH1蛋白在其他非肿瘤疾病如肩袖病[46]、糖尿病肾病[47]、吸烟相关动脉粥样硬化[48]等疾病的发展中具有一定作用,值得关注。总之,现在普遍认为,SASH1可能是一种具有抑癌功能的基因,但针对SASH1发挥抑癌作用的具体机制仍需深入探索。而且现有研究大部分为体外细胞培养得出的实验室结论,尚缺乏活体研究资料和临床资料。能否使SASH1作为治疗的靶点,其表达的程度和低表达时细胞微环境的变化能否通过药物进行控制,目前研究仍属空白。故进一步探究SASH1基因在人体肿瘤细胞中发挥作用的机制,可使研究者进一步开展有关肿瘤的发生发展机制的研究,为肿瘤的早期诊断和精确治疗提供指导数据。
作者声明:本文第一作者对于研究和撰写的论文出现的不端行为承担相应责任;
利益冲突:本文全部作者均认同文章无相关利益冲突;
学术不端:本文在初审、返修及出版前均通过中国知网(CNKI)科技期刊学术不端文献检测系统学术不端检测;
同行评议:经同行专家双盲外审,达到刊发要求。
