基于组合模型的转录调控网络构建算法研究*
2018-07-13刘晓燕张诚诚郭茂祖邢林林
刘晓燕,张诚诚,郭茂祖,邢林林
1.哈尔滨工业大学 计算机科学与技术学院,哈尔滨 150001
2.北京建筑大学 电气与信息工程学院,北京 100044
1 引言
转录调控是基因调控的主要过程,即转录因子通过结合位点进而控制目标基因的表达[1]。构建高精度的基因转录调控网络一直以来都是研究热点,其主要研究利用实验数据重构网络[2]。随着第二代大规模基因测序技术和高通量基因表达分析技术的发展,使得方便地获取基因表达数据、基因序列数据成为可能。此外,随着近年来对机器学习的不断研究,对构建基因转录调控网络的研究进入了一个新的阶段,构建高精度基因转录调控网络也成为可能。
转录调控的本质就是转录因子通过结合启动子位点,进而控制目标基因的转录水平来完成相应的功能。基因表达数据反映的是直接或间接测量得到的基因转录产物mRNA在样本中的丰度,这些数据可以用于分析哪些基因的表达发生了改变,基因之间有何相关性,在不同条件下基因的活动是如何受影响的。启动子是基因的一个组成部分,控制基因表达(转录)的起始时间和表达的程度。启动子本身并不控制基因活动,而是通过与转录因子结合,从而控制基因活动。
目前,构建基因转录调控网络的算法主要有如下几种。Bornholdt在2008年使用布尔网络模型构建调控网络,在网络中每个基因有开、关两个状态,状态“开”表示一个基因转录表达,形成基因产物,而状态“关”则代表一个基因未转录[3]。布尔网络虽然简单,但是建模能力有所欠缺。Werhli等人在2007年使用贝叶斯网络模型构建转录调控网络[4]。贝叶斯网络虽然可以建模比较复杂的网络,但是计算复杂度过高。Huynh-Thu等人在2011年使用基于树的模型 GENIE3(gene network inference with ensemble of trees)[5],将构建转录调控问题转换为机器学习里面的回归问题,在树节点分裂过程中得到基因与基因之间调控重要性的排名。2015年,Huynh-Thu等人提出了新的算法模型Jump3[6],对GENIE3算法进行了改进,之前的算法只用了基因表达数据,在新模型中Huynh-Thu引入了启动子状态这一变量,并且对树分裂节点标准进行了修改,从而带来了精度上的提升,但是计算时间复杂度过高,程序运行时间非常长,训练n个基因的转录调控网络Jump3算法至少需要n个子模型,即随机森林。Gillani[7]和Maetschke[8]等人在2014年将基因转录调控网络构建问题转为二分类问题,使用支持向量机进行训练,并比较了不同核函数的性能,实验中并未比较其他算法且只用了基因表达数据。Robiolo等人在2015年利用神经网络对转录调控网络进行建模[9],利用迭代次数、误差构建评分体系,进而计算基因之间的调控可能性,此方法同样存在计算复杂度过高的问题,得到n个基因的转录调控网络至少需要(n×(n-1))/2个神经网络模型。
本文主要研究的是转录调控网络的构建算法。一方面,目前研究方法只用了基因表达数据;另一方面,目前对于基因功能研究方面已经形成了比较完善的GO注释。GeneOntology(基因本体)是一种用于功能注释的树型结构数据[10],具有相同GO注释的基因功能往往相似。于是本文利用基因表达数据、基因序列数据、基因注释数据这3个数据来源。此外,综合考虑精度和计算复杂度,本文将转录调控网络构建问题转化为二分类问题,提出了基于深度自编码器和组合模型构建基因转录调控网络。
本文组织结构如下:第2章介绍了数据获取、数据预处理以及负例集的构建。第3章详细阐述了本文提出的基于深度自编码器的XGBoost和逻辑回归组合模型DAXL(combined model with XGBoost and logistic regression based on deep AutoEncoder)。第 4章以拟南芥调控网络的构建为例,对DAXL方法进行了验证,并与GENIE方法进行了对比,给出了实验结果。第5章对全文进行总结。
2 数据获取、预处理及负例集的构建
2.1 数据获取
转录因子活性会明显影响目标基因的表达水平,因此两者在转录水平上的表达谱具有相关性[11]。本文从GEO(gene expression omnibus)上获取编号为GSE41212[12]的基因表达数据,该数据集从时间和空间两个角度详细分析了拟南芥种子发芽的过程,记录了拟南芥种子的4个组织在9个时间点的基因表达值。基因序列数据主要包括两部分:启动子序列数据、蛋白质的氨基酸序列数据。其中启动子的序列数据是从TAIR上下载,氨基酸序列数据是从http://www.arabidopsis.org/上下载。本文的GO数据可以从http://geneontology.org/page/download-annotations上下载。正例集是已经被实验验证的,确定有基因转录调控关系的转录因子、目标基因对,也是后续进行模型训练必要的数据。AGRIS收录了19 013对具有直接调控关系的转录因子和目标基因。本文选取该数据集作为正例集,可从http://arabidopsis.med.ohiostate.edu/上下载。
2.2 数据预处理
本文收集的生物数据有两个主要问题:一是基因序列数据、基因注释数据都是字符串,无法直接进行模型训练;二是把基因表达数据、基因序列数据、基因注释数据以基因ID作为主键进行链接时存在缺失数据。
本文获得基因表达数据、基因序列数据、基因注释数据的具体处理如下。
从GEO上获取的基因表达数据是数值型的,在与基因序列数据、基因注释数据进行以基因ID为主键的链接时,存在部分缺失的情况。因为模型中包括集成树模型,对缺失值不敏感,所以本文直接用-1填充缺失值,从而得到116维特征:

在生物学中,相邻的3个核苷酸被称为密码子,每个密码子编码一个氨基酸,因为对于启动子序列数据而言,每一条启动子序列,都可以得到全部密码子的频次,对于每一个转录因子都可以得到如下64维特征:

蛋白质序列可以表示成由20个氨基酸字符组成的字符串,其字符集合为{A,V,L,I,F,P,M,S,T,C,W,Y,N,Q,D,E,K,R,H,G},对于每一条蛋白质序列数据,统计这20个字符的出现频次,从而得到如下的20维特征:
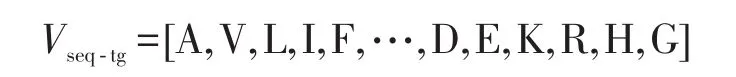
转录因子通过调控目标基因参与同一生物过程,因此两者往往具有相同的功能。转录因子在基因本体(GO)数据库中,每个GO ID是一个唯一的标识符,对应某一术语名称。因此,可以利用GO术语建立特征向量[13]。本文利用拟南芥的GO数据,最终大约取得3 112维特征。具体步骤如下:
(1)根据选定的数据集中的基因ID,提取对应每一个ID的所有GO术语。
(2)将提取出的GO术语依次编号。
(3)假设共有n个GO术语,若基因含有i(i=1,2,…,n)号GO术语,则为1,否则为0,则每个基因功能信息可以表示成一个n维向量:
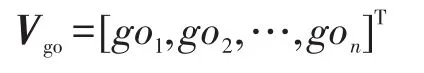
通过数据预处理,对于每一对转录因子、目标基因,可以得到如下一条向量:
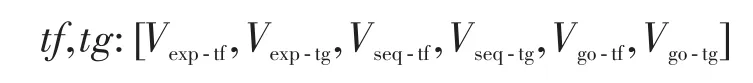
2.3 构建负例集
将转录调控网络问题转换为机器学习二分类问题,需要构建负样本。本文从生物背景出发,构建负样本采用Yu等人在文献[14]中提出的方法。构建负样本的基本思想是:假设基因为G,转录因子为T,如果基因G不包含转录因子T的任意一个结合位点,则认为(T,G)这一对基因为负样本。
3 基于深度自编码器的XGBoost、逻辑回归组合模型DAXL
首先使用深度自编码器训练原始高维基因注释数据,得到新的低维可以表征基因注释数据的向量;然后把基因表达数据、基因序列数据、新的向量交给XGBoost进行训练,对于训练好的XGBoost模型,可以得到每一条样本最终落在每一个树上叶子节点的信息,从而进行01编码,即样本落在某一个叶子节点上为1,否则为0;最后将01编码向量交给逻辑回归训练进行分类,最终得到转录调控网络。整体算法流程如图1所示。
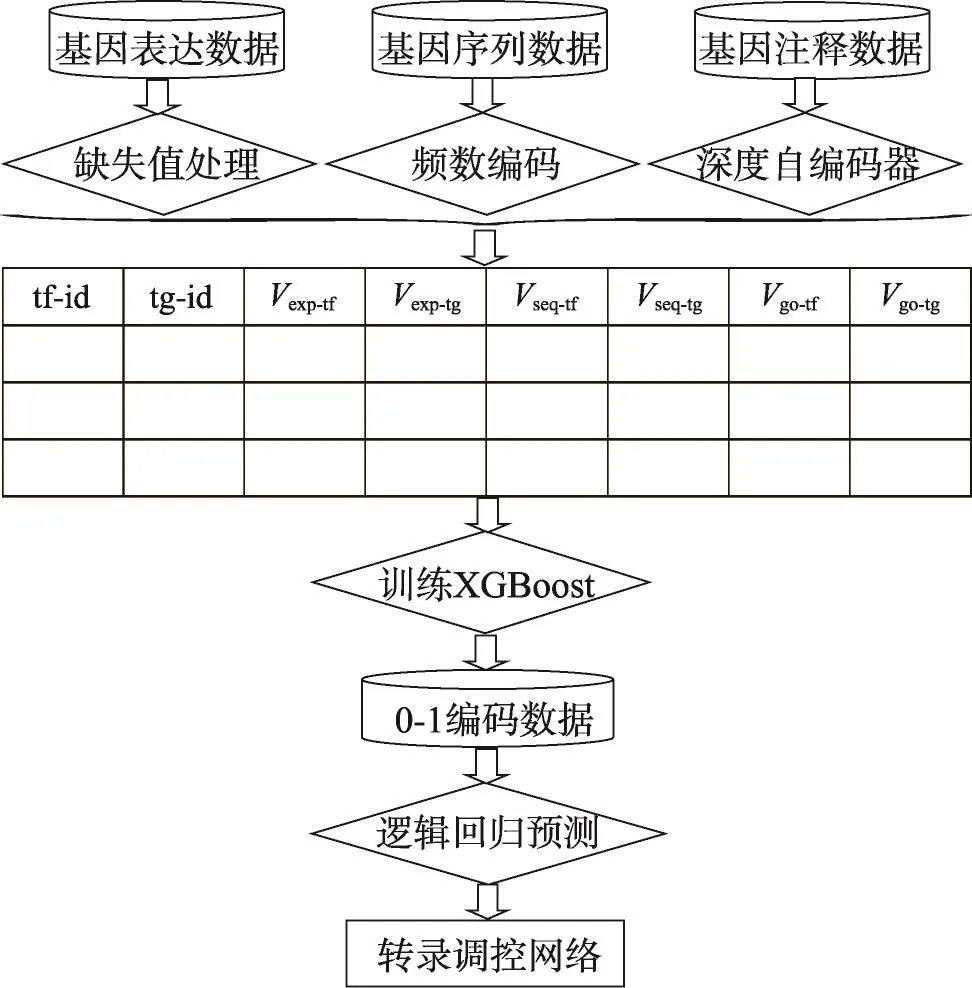
Fig.1 Flow chart of DAXL图1 DAXL整体流程图
3.1 基于深度自编码器的基因注释数据降维方法
经过数据预处理的特征有6 540维,其中基因注释数据特征特别稀疏,不利于基于集成树的模型进行训练。为了解决这个问题,本文利用深度自编码器[15-16]进行Embedding,即先训练深度自编码器模型,然后对于训练好的模型直接取其中隐层作为新的输入样本。
深度自动编码器即多层自编码器,其输入输出一致,内部经过多层的编码、解码,具有很好的学习能力。本文使用深度自编码器完成高维特征的embedding目标。利用深度自编码器隐层单元数量可以自行定义的特性,将隐层节点设置为从高到低,再从低到高。然后提取最中间隐层学习到的向量作为原始输入,如图2所示,进而解决基因注释数据高维且稀疏的问题。
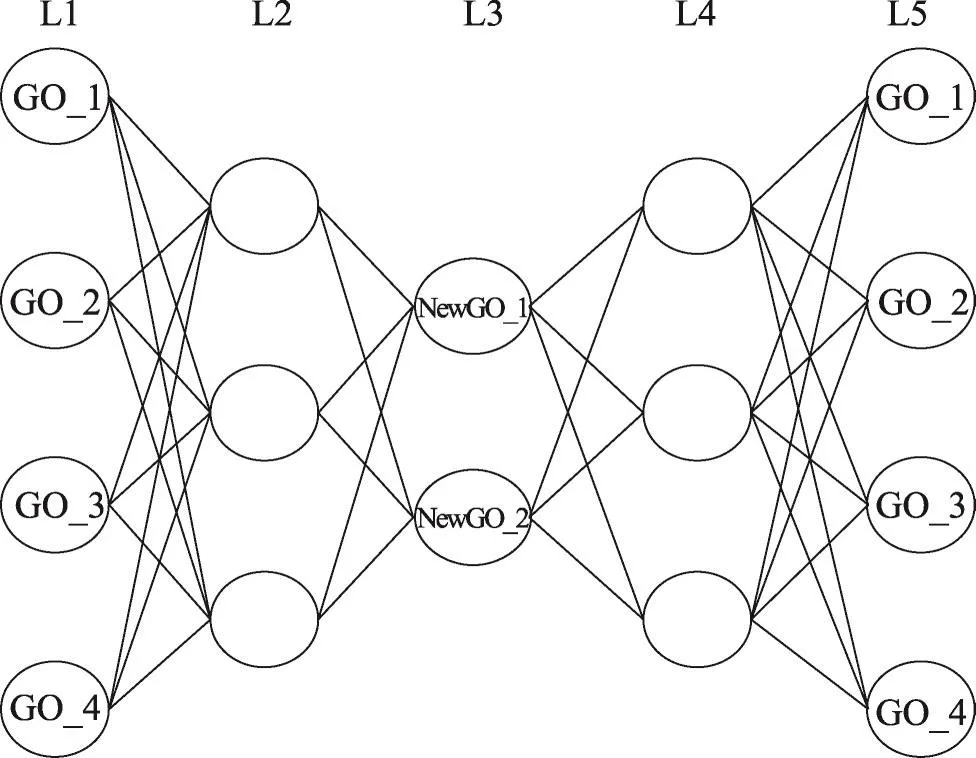
Fig.2 DeepAutoEncoder图2 深度自编码器
如图2所示,本文使用五层的深度自编码器,第一层(L1)和第五层(L5)是相同的向量,中间有3个隐藏层。以第一层与第五层向量差的平方作为学习目标,使用反向传播算法学习参数,通过多轮迭代后,得到最终的网络结构。本文为了获得可以表征原始输入向量的低维向量,用第三层(L3)的输出表示原始输入向量。
3.2 基于XGBoost的01编码器
XGBoost模型最早在2014年由Chen等人提出[17],它是boosting的一个扩展变体。XGBoost在以决策树为基础的基学习器构建boosting集成的基础上,使用泰勒展开式近似目标函数,利用一阶、二阶导数,从而提高精度。从图3中可以看出,XGBoost由多个弱分类器组成,且每一分类器的学习都相互独立,比较容易进行数据并行、特征并行,因此并行粒度大。此外,树模型是一个典型的if-else结构,可以处理非线性特征问题。
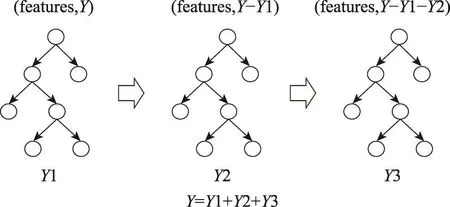
Fig.3 Schematic diagram of XGBoost图3 XGBoost示意图
3.3 基于逻辑回归的转录调控关系预测方法
逻辑回归模型(logistic regression)是最大熵模型的一个特例[18],它的雏形是线性回归,是一个线性模型,不可以直接处理非线性问题。通过sigmoid函数进行转换,将预测值映射到[0,1],从而转变为分类模型,比较适合01编码训练数据。可以通过极大似然估计、梯度下降得到相对较优解。从图4中可以看出,逻辑回归可以通过行并行、列并行的方式计算梯度。
3.4 DAXL算法
为了充分发挥非线性、线性模型的优势,借鉴Facebook 2014年在计算广告领域提出的新模型[19],本文构建了组合模型,如图5所示。
样本X先通过XGBoost进行训练,XGBoost训练完成后,对于每一条输入向量,可以根据叶子节点的信息得到新的一维向量。举例来说,在图5中,训练好的XGBoost模型有两棵树,第一棵树有3个叶子节点,第二棵树有2个叶子节点。如果一条样本在XGBoost模型的第一棵树上落在第一个叶子节点,在XGBoost模型的第二棵树上落在第一个叶子节点,在0-1 Code层可以得到(1,0,0,1,0)这个新向量,然后这个向量再通过逻辑回归模型进行训练。
4 实验
4.1 01编码有效性验证
用TSNE(t-distributed stochastic neighbor embedding)[20]将高维向量映射到二维空间,如图6、图7所示。图6是原始基因表达数据,图7是经过01编码之后得到的新的数据。其中红色的点是正例,蓝色的点是负例,从而可以看出经过01编码之后,数据集正负例集相对分开,更加利于模型进行分类。
4.2 深度自编码器中间层参数对结果的影响
原始基因注释数据经过离散化后有3 112维,深度自编码器在一定程度上可以降维且可以学习到一个更好的表达形式,从而提高后续组合模型的精度。图8展示了深度自编码器中间层节点数在10、50、100、200、500时对XGBoost、XGBoost+LR最终预测结果F1值的影响,其中节点数为0时表示没有使用深度自编码器对基因注释数据进行处理。一方面说明组合模型DAXL精度比XGBoost高;另一方面说明深度自编码器可以对离散化的基因注释进行充分学习,从而得到更好的表达形式。
4.3 转录调控网络构建结果
由于构建负例集时存在假阳性问题,即实际是正例可能被认为是负例进行训练,本文使用F1作为评价指标。F1评价指标综合考虑准确率P和召回率R[21]。
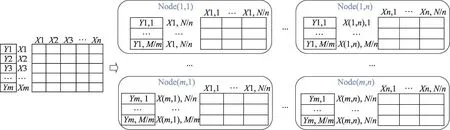
Fig.4 Logistic regression图4 逻辑回归

Fig.5 Combined model图5 组合模型
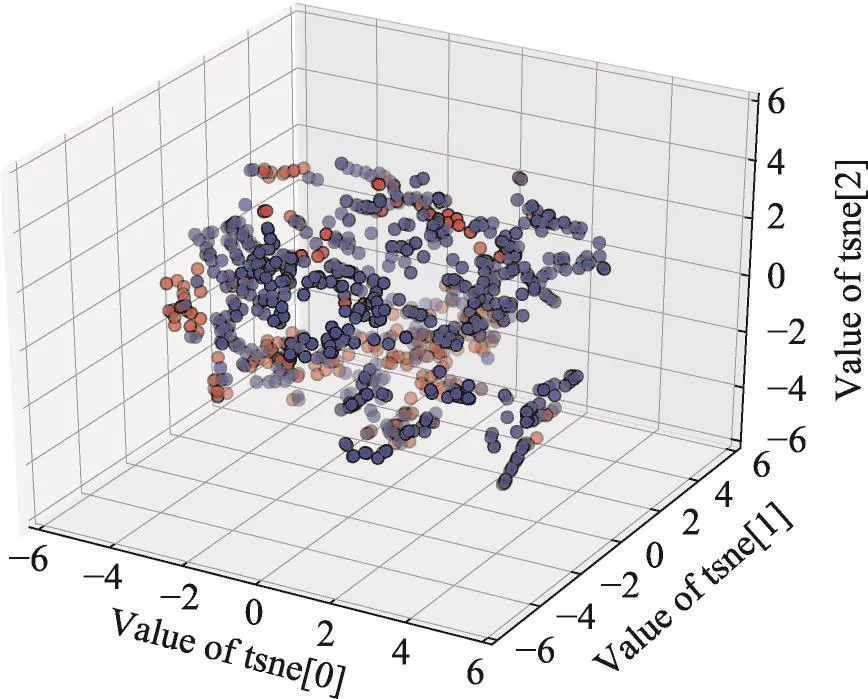
Fig.6 Initial image based on TSNE图6 原始TSNE图
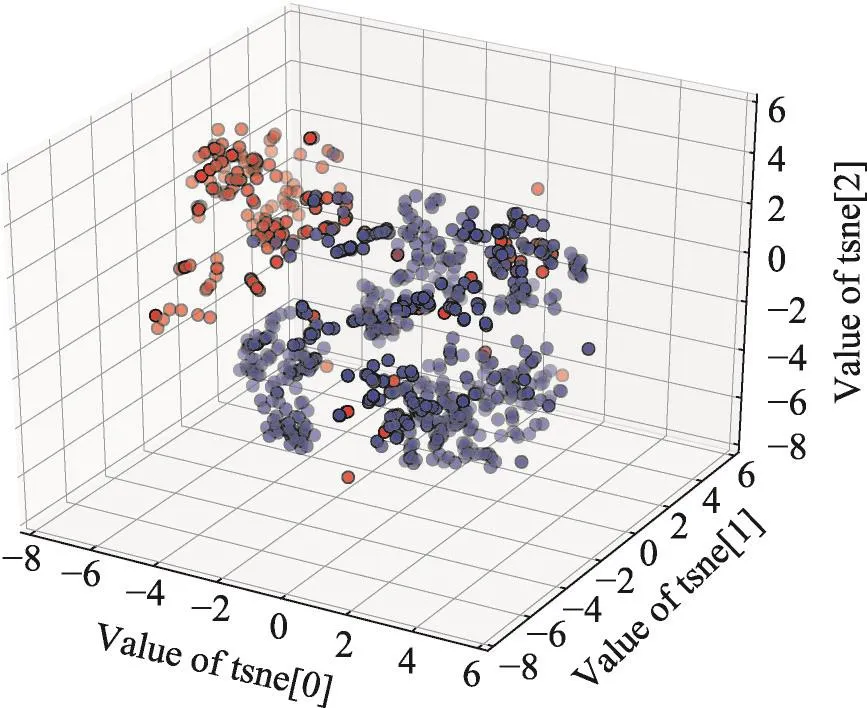
Fig.7 0-1 code image based on TSNE图7 0-1编码TSNE图
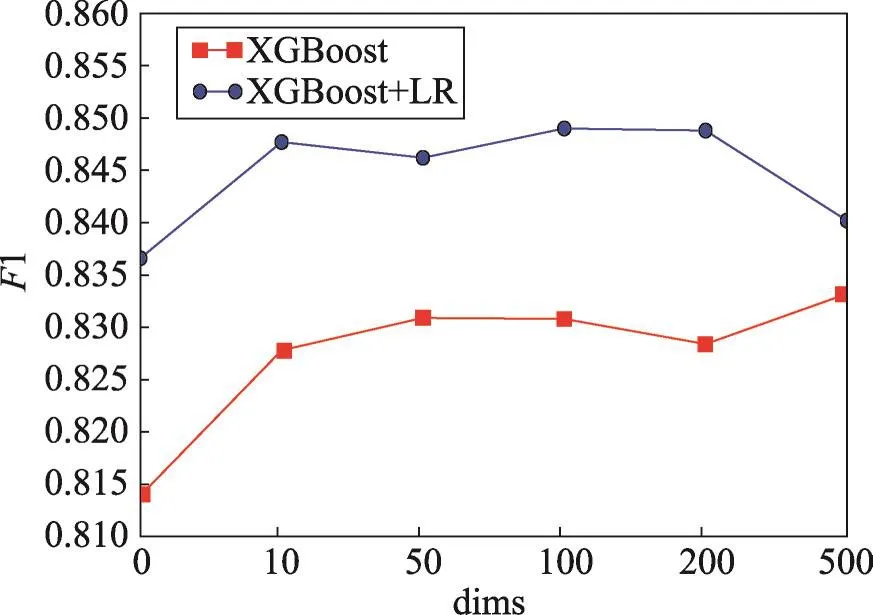
Fig.8 Influence of the number of nodes in inter layer of deepAutoEncoder on XGBoost and XGBoost+LR图8 深度自编码器中间层节点数对XGBoost、XGBoost+LR的影响
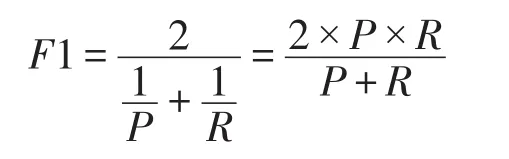
从表1中可以看出,XGBoost模型比逻辑回归模型精度高,因为XGBoost具有非线性拟合能力。DAXL模型的精度比XGBoost高,因为采用了01编码,同时综合了XGBoost、逻辑回归模型的优点。

Table 1 Result of experiments inArabidopsis dataset表1 在拟南芥数据集上的实验结果
从表2中可以看出,DAXL方法比只使用基因表达数据的GENIE3方法好。同时,在相同的数据集上,DAXL模型也比支持向量机方法好,因为支持向量机对缺失值敏感,且核函数的选择对精度影响很大。

Table 2 Result of experiments inArabidopsis dataset表2 在拟南芥数据集上的实验结果
4.4 预测新的转录调控关系
为了进一步验证本文算法的可靠性以及本文工作的意义,从http://arabidopsis.med.ohio-sta te.edu/得到最新的转录调控关系,其中有69对转录调控关系在原来的训练验证过程中没有出现过,本文使用DAXL模型对这69对调控关系进行预测。如图9所示,发现有62对的预测概率在0.5以上,有47对的预测概率在0.8以上。
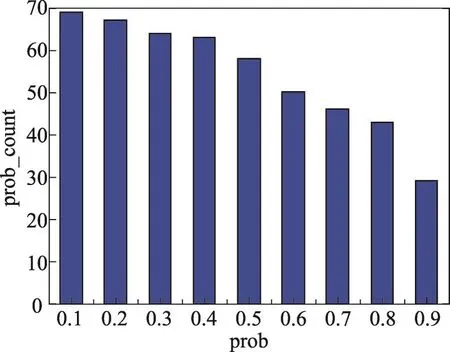
Fig.9 Predictive probability of novel transcriptional regulatory relationships图9 新的转录调控关系预测概率
5 总结
本文分析了目前经典的基因转录调控网络构建算法,针对存在的问题,提出了基于深度自编码器和组合模型的方法。本文方法充分利用了基因表达数据、基因序列数据、基因注释数据,用深度自编码器巧妙地解决了基因注释数据高维稀疏的问题;使用XGBoost与逻辑回归的组合模型,结合生物背景知识,把基因转录调控网络构建问题转为二分类问题,从而构建了基因转录调控网络。本文在真实的拟南芥数据集上对提出的方法进行了验证,并与现有方法进行了比较。结果说明本文方法比单一采用LR、XGBoost更加有效,较对比方法GENIE3提高了15个百分点。
