亲水作用液相色谱-串联质谱测定烟叶中马来酰肼及其糖苷
2018-07-10师君丽邓小鹏孔光辉卢秀萍
逄 涛, 林 茜, 李 勇*, 师君丽, 邓小鹏,孔光辉, 卢秀萍, 晋 艳
(1. 云南省烟草农业科学研究院, 云南 玉溪 653100; 2. 玉溪师范学院, 云南 玉溪 653100)
马来酰肼(1,2-二氢-3,6-哒嗪二酮,maleic hydrazide, MH)是一种人工合成的植物生长调节剂,从20世纪40年代开始应用于农作物[1],是国内外烟草行业常用的抑芽剂。马来酰肼对动物代谢的影响研究未获得确定的结论,部分研究认为马来酰肼对哺乳动物毒性较低[2,3],而Swietlińska等[4]的研究则认为其对小鼠和大鼠具有一定的致癌性,因此人类接触马来酰肼有一定的风险。联合国粮农组织(FAO)对洋葱(球)、土豆(块茎)、大蒜(头)等作物中的马来酰肼设定了最大限量值[5]。国际烟草科学研究合作中心(CORESTA)在2016年颁布的烤后烟叶农残指导残留水平文件中规定了马来酰肼(自由态和糖苷结合态)的残留水平为80 mg/kg[6]。同样,中国烟草总公司也于2014年公布了包括马来酰肼在内的烟叶农药的最大残留要求[7]。因此建立一种简单、快速的烟叶中马来酰肼残留分析方法对于保障烟叶安全、杜绝马来酰肼抑芽剂的滥用具有重要意义。
马来酰肼在烟叶中以自由态及糖苷结合态存在。其中糖苷结合态马来酰肼包括马来酰肼-O-β-D-葡萄糖苷[8]和马来酰肼-N-β-D-葡萄糖苷[9]。近年来,人们对烟叶中农药残留检测技术研究日趋重视[10-12],其中液相色谱法[13]和液相色谱-质谱法[14,15]是目前报道较多的马来酰肼测定方法。这些方法包含只测定自由态马来酰肼和将马来酰肼糖苷水解后测定其总量两种类型。但是,只测定自由态马来酰肼无法反映马来酰肼的真实残留量,而将马来酰肼糖苷水解后测定其总量的方法目前无法对两种糖苷物质是否彻底水解进行评价,且采用强酸强碱(浓盐酸和氢氧化钠)进行样品处理对实验操作者带来操作风险,也对分析仪器带来被腐蚀的风险。
本研究根据Newsome方法[16]合成了马来酰肼-O-β-D-葡萄糖苷,并以此为基础在喷洒了马来酰肼的烟叶中鉴定出马来酰肼-O-β-D-葡萄糖苷和马来酰肼-N-β-D-葡萄糖苷(见图1)。通过前处理方法的筛选、色谱-质谱方法的优化,建立了烟叶中马来酰肼及其糖苷的定量分析方法。方法前处理步骤简单,避开了传统方法中的水解反应和固相萃取步骤,分析时间短,灵敏度高,方法的建立为快速、准确监控烟叶中的马来酰肼残留提供了一种新的技术路线。
1 实验部分
1.1 仪器与试剂
甲醇(色谱级)购于Merck公司,马来酰肼购于Sigma公司(北京),马来酰肼-O-β-D-葡萄糖苷采用Newsome方法[16]合成,纯度为82.04%。氧化银、硫酸钙、2,3,4,6-四乙酰氧基-α-D-吡喃葡萄糖溴化物、二氯甲烷、硅酸、甲醇钠等(用于合成马来酰肼-O-β-D-葡萄糖苷)购于北京百灵威公司。
Waters Acquity UPLC液相色谱仪(Waters公司,美国), Absciex 5500三重四极杆质谱(Sciex公司,加拿大); Milli-Q纯水机(Millipore公司,美国); Eppendorf 5804高速离心机(Eppendorf公司,德国), Talboys数显型多管式涡旋混合器(安谱科学仪器公司,上海); CP2245分析天平(感量0.000 1 g, Sartorious公司,德国)。Christ Alpha 1-2 LD冻干机(Martin Christ公司,德国)。
1.2 烟叶样品的制备
用于实验的栽培烟草(K326)种植于云南省烟草农业科学研究院玉溪研和实验基地,移栽3个月后开始马来酰肼喷洒实验。40株实验烟草被分成马来酰肼喷洒组(25株)、空白对照喷洒组(10株)和空白组(5株)。其中马来酰肼喷洒组每株喷施20 mL 10 g/L的马来酰肼(溶于含3%(体积分数)三乙醇胺的水中,三乙醇胺用于促进马来酰肼溶解),空白对照喷洒组每株喷施20 mL的空白溶液(含3%(体积分数)三乙醇胺的水溶液),空白组烟株未做处理。喷洒实验后,分别于第0、7、14、21、28天采集处理的烟株烟叶。其中马来酰肼喷洒组和空白对照组每次分别采收5株和2株烟叶。第0天采收烟叶时间为喷洒后约半小时,此时烟叶上喷洒的试剂已不可见。空白组(5株)烟叶在第28天集中采收,并混合成一个空白样品。所采集的烟叶一部分直接置于-80 ℃冰箱保存,另一部分采用三段式烘烤方法[17]进行烘烤后常温保存。
1.3 样品前处理
新鲜烟叶样品制备:将新鲜的烟叶样品放入研钵(或低温自动研磨仪),加液氮冷冻,研磨粉碎(小于40目)。磨好的样品迅速转入离心管封存于-80 ℃冰箱。
烤后烟叶样品制备:将烤后烟叶样品放入研钵,研磨粉碎(小于40目)。磨好的样品转入离心管封存于4 ℃冰箱。
本实验所建立的样品前处理方法:准确称取100 mg液氮下粉碎的新鲜烟末(或10 mg烤后烟叶粉末)于1.5 mL离心管,依次加入350 μL乙腈、500 μL甲基叔丁基醚、650 μL水,超声20 min,离心(转速10 000 r/min)5 min,取下层清液100 μL,离心浓缩(真空度0.018 Pa)30 min去掉溶剂,加入100 μL乙腈复溶,复溶液转入带内衬的液相色谱进样小瓶,待分析。
水解后测马来酰肼总量法的前处理方法[18]:称取5 g烟草粉末于250 mL磨口圆底烧瓶中,加入50 mL 4 mol/L的盐酸溶液。将圆底烧瓶放入电加热套中,装上球形冷凝管,加热回流。控制回流速度使蒸汽升至球形冷凝管第二球处,连续加热回流1 h。回流完毕放至室温后用定性滤纸过滤试样,滤液收集至100 mL容量瓶中,用适量4 mol/L的盐酸溶液淋洗冷凝管和萃取过的试样,洗液均经过滤合并至该100 mL容量瓶中,并用盐酸溶液定容。取2 mL甲醇和2 mL 0.1 mol/L氢氧化钠溶液依次预淋洗C18固相萃取小柱。从100 mL容量瓶中准确移取2 mL提取液加到C18固相萃取小柱上,并即刻开始收集流出组分至5 mL容量瓶中,用2 mL 0.1 mol/L氢氧化钠溶液洗脱,合并洗脱液至该5 mL容量瓶中,并用0.1 mol/L氢氧化钠溶液定容,定容液经0.45 μm水相滤膜过滤后备用。
QuEChERS法:见文献[19]。
进行样品前处理方法对比时,3种前处理方法得到的马来酰肼均稀释至相同的浓度。为了便于亲水作用色谱分析,3种方法处理最终溶剂均置换成乙腈。
1.4 液相色谱-质谱条件
液相色谱条件色谱柱为Waters Acquity UPLC@BEH HILIC (100 mm×2.1 mm, 1.7 μm)。流动相:A为含0.2%(v/v)甲酸的水溶液,B为含0.2%(v/v)甲酸的乙腈溶液。梯度洗脱条件:0~1.00 min, 2%A; 1.00~4.30 min, 2%A~12%A; 4.30~4.31 min, 12%A~50%A; 4.31~6.00 min, 50%A; 6.00~6.01 min, 50%A~2%A; 6.01~7.00 min, 2%A。流动相流速为0.25 mL/min;柱温23 ℃;进样量5 μL。
质谱条件多反应监测,正离子模式采集。离子源参数如下:在液相色谱保留时间0~2.5 min内:喷雾电压4 000 V;气帘气172 kPa;碰撞气62 kPa;温度700 ℃;离子源气体1为276 kPa;离子源气体2为621 kPa;去簇电压60 V;入口电压10 V;碰撞池外部电压15 V。在液相色谱保留时间2.5~7.0 min内:喷雾电压5 500 V;气帘气172 kPa;碰撞气34 kPa;温度250 ℃;离子源气体1为276 kPa;离子源气体2为414 kPa;去簇电压60 V;入口电压10 V;碰撞池外部电压15 V。马来酰肼及其糖苷的色谱-质谱条件见表1。
1.5 标准曲线绘制和定量计算
基质添加标准曲线的绘制:以甲醇为溶剂,配制10 mg/L的马来酰肼及马来酰肼-O-β-D-葡萄糖苷混合标准溶液。分别移取5、10、20、40、80、150 μL混合标准溶液至1.5 mL离心管,再分别称取10 mg空白烟末(不含马来酰肼及其糖苷)至离心管作为样品基质,制成5、10、20、40、80、150 mg/kg含量梯度的待测样本。离心浓缩,除去样品中的甲醇溶剂。将制好的样本按照1.3节样品前处理和1.4节色谱-质谱条件部分所述方法进行处理,得到每个含量梯度样本对应的色谱-质谱峰面积。将得到的峰面积与其对应的含量梯度进行线性拟合,得到校准曲线拟合方程和线性相关系数。
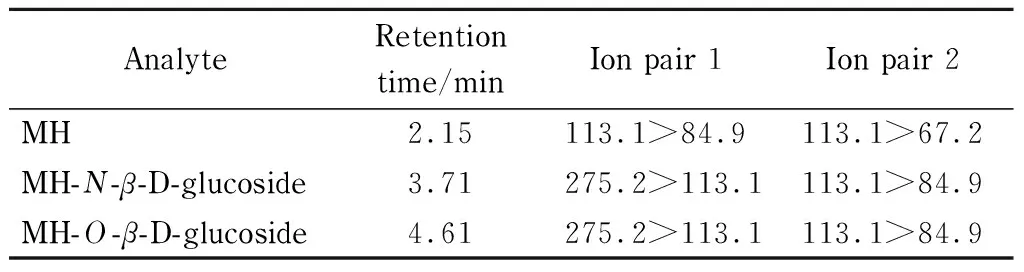
表 1 马来酰肼及其糖苷分析的色谱-质谱分析条件
溶剂空白标准曲线的绘制:除不添加空白烟末以外,其余步骤与基质添加标准曲线的绘制相同。
将得到的实际样品中的马来酰肼及其糖苷的色谱-质谱峰面积输入校准曲线方程,计算得到相对应的分析物含量。由于无法获得马来酰肼-N-β-D-葡萄糖苷的标准物质,且马来酰肼-N-β-D-葡萄糖苷与马来酰肼-O-β-D-葡萄糖苷互为位置异构体,本方法中具有相同的MRM质谱离子,因此采用马来酰肼-O-β-D-葡萄糖苷的校准曲线对马来酰肼-N-β-D-葡萄糖苷进行定量。烟叶中马来酰肼(游离态和糖苷结合态)的含量计算公式见式(1)。
CMH=CMH-F+CMH-G×MMH/MMH-G
(1)
其中,CMH为烟叶中马来酰肼(自由态和糖苷结合态)的含量(mg/kg),CMH-F和CMH-G分别为自由态和糖苷结合态马来酰肼的含量(mg/kg),MMH和MMH-G分别为马来酰肼和马来酰肼-葡萄糖苷的相对分子质量。
1.6 基质效应评价
通过基质添加标准曲线和溶剂空白标准曲线的斜率评价基质效应。具体计算公式见式(2)[20]。
ME=(1-X2/X1)×100%
(2)
其中,ME代表基质效应,X1和X2分别代表溶剂空白标准曲线和基质添加标准曲线的斜率。
2 结果与讨论
2.1 烟叶样品中马来酰肼-葡萄糖苷的确定
Towers和Hutchinson[21]通过14C同位素标记的方法证实了喷洒马来酰肼的小麦叶片产生了2个马来酰肼葡萄糖结合物。Frear等[8]以及Tagawa等[9]随后在烟草实验中证实了这2个葡萄糖结合物为马来酰肼-O-β-D-葡萄糖苷和马来酰肼-N-β-D-葡萄糖苷。虽然这两个糖苷物质已经被鉴定出来,但由于相关标准物质和质谱图的缺乏,色谱-质谱分析中这两种糖苷的保留时间及特征离子有待确认。
本研究采用Newsome方法[16]合成了马来酰肼-O-β-D-葡萄糖苷,通过在质谱负离子模式下的分子离子m/z273 [M-H]-、二级碎片m/z111[M-162-H]-以及中性丢失m/z162(葡萄糖基),证实了马来酰肼-葡萄糖苷的合成。Newsome[16]已经通过核磁共振光谱等技术确认了该方法合成的糖苷为马来酰肼-O-β-D-葡萄糖苷,并确认喷洒了马来酰肼的烟叶中产生的马来酰肼的主要糖苷为马来酰肼-O-β-D-葡萄糖。本实验在确认马来酰肼-葡萄糖苷合成成功以后,通过对比合成的糖苷与喷洒了马来酰肼烟叶中主要马来酰肼糖苷色谱峰在色谱保留时间(4.61 min)和MRM质谱离子对(275.2>113.1和113.1>84.9,正离子模式)的完全一致,确认了合成的物质为马来酰肼-O-β-D-葡萄糖苷。合成的马来酰肼-O-β-D-葡萄糖苷经气相色谱-质谱(全扫模式)确定其纯度为82.04%(见图2)。

图 2 本实验合成的马来酰肼-O-β-D-葡萄糖苷的气相色谱-质谱全扫描色谱图Fig. 2 GC-MS full scan chromatogram of synthesized MH-O-β-D-glucoside GC conditions: column, DB-5MS (30 m×0.25 mm×0.25 μm); temperature program: 0-1 min, 60 ℃; 1-45 min, 60-280 ℃; 45-60 min, 280 ℃.
通过假设马来酰肼-N-β-D-葡萄糖苷与马来酰肼-O-β-D-葡萄糖苷具有相同的MRM离子对和接近的保留时间(因为两者为位置异构体),在色谱保留时间为3.71 min的位置找到了马来酰肼-N-β-D-葡萄糖苷的候选物,通过对比马来酰肼喷洒后的烟叶样品和空白样品,发现该候选物只存在于马来酰肼喷洒后的烟叶样品中,且其色谱峰面积只有相同样品中马来酰肼-O-β-D-葡萄糖苷峰面积的5%~10%,与文献[21]报道的马来酰肼两个糖苷的相对含量关系一致,综合以上信息确定该候选物为马来酰肼-N-β-D-葡萄糖苷。
2.2 样品前处理方法的筛选
研究已证明马来酰肼施用于烟草(或其他作物)后会生成马来酰肼-O-β-D-葡萄糖和马来酰肼-N-β-D-葡萄糖[8,9],由于目前尚无这两种糖苷的标准品可购买,文献[18]报道的马来酰肼残留检测方法通常先采用盐酸将糖苷水解,然后再用色谱法测定马来酰肼的总量(即水解测总量法)。本实验人工合成了马来酰肼-O-β-D-葡萄糖,在此基础上从喷洒了马来酰肼的烟叶中鉴定出马来酰肼-O-β-D-葡萄糖和马来酰肼-N-β-D-葡萄糖。因此本实验无需对糖苷进行水解即可直接测定马来酰肼及其两个糖苷(即直接测定法)。
本实验对比分析了水解测总量法、直接测定法以及多种农药残留测定最常用的QuEChERS法[19]对喷洒马来酰肼27天后采集的烟叶样品中马来酰肼及其糖苷的提取和纯化效果(图3a),结果发现QuEChERS法的提取效果最差,提取物中未见马来酰肼-O-β-D-葡萄糖苷和马来酰肼-N-β-D-葡萄糖苷,马来酰肼的峰也很弱,几乎要被基线掩盖。水解测总量法和本实验建立的直接测定法均能获得较明显的马来酰肼色谱峰,且直接测定法马来酰肼色谱峰的信号稍强于水解测总量法,故初步判断直接测定法较好。

图 3 不同样品前处理方法的效果比较Fig. 3 Comparison of different sample preparation methods a. a tobacco leaf sample on the 28th day after MH spraying; b. a blank tobacco sample; c. a blank tobacco leaf sample spiked with 1 mg/kg maleic hydrazide.
为了进一步比较水解测总量法和直接测定法,对比分析了两种方法处理空白基质和其添加1 mg/kg马来酰肼时的色谱响应情况,结果显示,直接测定法的空白基质提取液响应信号与水解测总量法相当(见图3b),但在空白基质中添加1 mg/kg马来酰肼时直接测定法提取物的马来酰肼色谱峰的响应信号明显强于水解测总量法(见图3c)。由于直接测定法的前处理方法仅包括萃取、离心、溶剂置换等步骤(总时间约1.5 h),较水解测总量法(包括水解反应、固相萃取、2次定容等步骤,总时间超过4 h)显著简化,且本实验所建立的方法不受水解反应装置限制,较水解测总量法具有更大的处理通量,因此最终采用本实验建立的直接测定法。
2.3 色谱条件的筛选
文献报道的基于液相色谱的马来酰肼分离方法主要有反相色谱柱分离法[13]和多孔石墨碳色谱柱分离法[14,15]。反相色谱法是马来酰肼分离使用较多的方法,但马来酰肼在反相柱上保留较弱,分离效果不佳[14]。陈晓水等[14]和Wang等[15]认为,多孔石墨碳色谱柱对烟叶中的马来酰肼分离效果较好,但这2例多孔石墨碳色谱柱分析方法的分离时间分别达到30 min[14]和35 min[15]。本实验采用细粒径(1.7 μm)的亲水相互作用色谱柱(HILIC)在7 min内实现了马来酰肼及其两个糖苷的完全分离(见图4),大大缩短了分析时间,并提高了分析灵敏度(色谱峰更窄)。
2.4 质谱条件的优化
实验发现,马来酰肼的响应强度随离子源温度的升高而升高,马来酰肼-O-β-D-葡萄糖苷的响应强度在250 ℃达到最高,马来酰肼-N-β-D-葡萄糖苷的响应强度在250 ℃达到较高的水平,随后在250~550 ℃之间变化较小(见图5)。马来酰肼-O-β-D-葡萄糖苷在温度大于250 ℃时响应迅速下降,其原因是在较高温度下它会发生较大的离子源源内裂解,失去葡萄糖基,产生大量的[M-162+H]+离子。因此,本实验针对马来酰肼和其糖苷分别优化了离子源参数,得到马来酰肼和其糖苷的最佳离子源温度分别为700 ℃和250 ℃。同时实验还对其他离子源参数做了相应的优化,结果见1.4节。
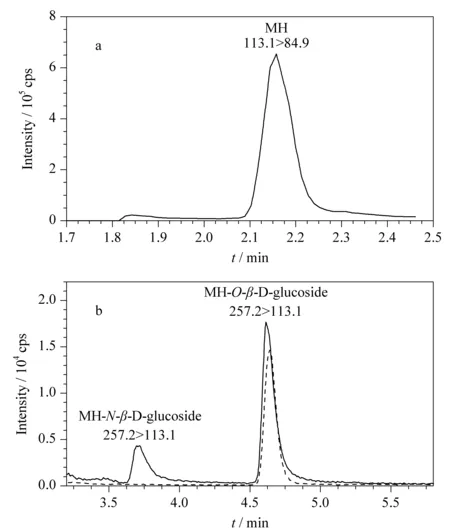
图 4 烟叶中马来酰肼及其糖苷的色谱图Fig. 4 Chromatograms of MH and its glucosides in tobacco leaf a. spray voltage, 4000 V; source temperature, 700 ℃. b. spray voltage, 5500 V; source temperature, 250 ℃. Dotted line: chromatogram of MH-O-β-D-glucoside synthesized using the Newsome method[16].

AnalyteMatrixStandard curver2Repeatabilities*/%Intra-dayInter-dayMHnoney=55031x-1572050.99633.47.9tobacco leavesy=11770x-267300.99722.78.3MH-O-β-D-glucosidenoney=46146x-1869590.99722.55.6tobacco leavesy=6363.6x-8554.30.99713.87.1
y: peak area;x: content, mg/kg. * Intra-day and inter-day repeatabilities: relative standard deviations of triplicate detection in one day and three consecutive days, respectively, at spiked level of 40 mg/kg.
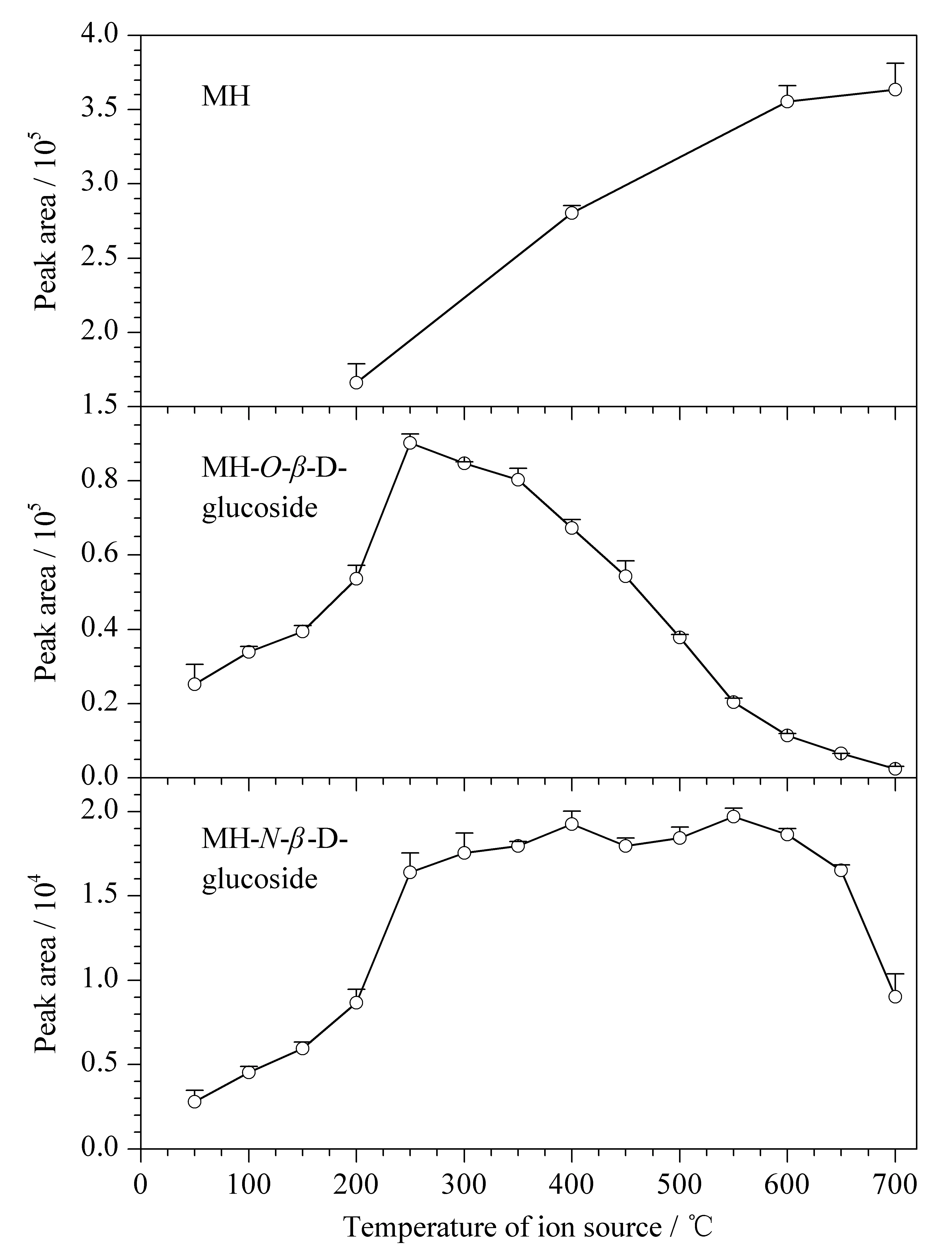
图 5 马来酰肼及其糖苷的响应随离子源温度的变化曲线Fig. 5 Temperature-response curve of MH and its glucosides
2.5 方法评价
从基质效应、重复性、回收率、定量限、检出限等方面评价了所建立的分析方法。结果发现,烟叶基质对马来酰肼及马来酰肼-O-β-D-葡萄糖苷的基质效应分别为78.6%和86.2%(根据1.6节所述方法和表2校准曲线数据计算),可见烟叶基质对马来酰肼及其糖苷的响应强度产生了很大的抑制作用,但基质添加标准曲线仍可获得与溶剂空白标准曲线相似的线性响应(r2,见表2)。因此本实验采用基质添加标准曲线对目标分析物进行定量,以避免基质效应对定量结果的影响。
向空白烟叶样品中分别添加10、40、80 mg/kg的马来酰肼和马来酰肼-O-β-D-葡萄糖苷进行加标回收试验,并同时测定日内重复性和日间重复性。结果表明方法的重复性(见表2)和回收率(见表3)均符合欧盟委员会“食品和饲料中农残分析的质量控制和方法评价指导文件”[22]的相关要求(回收率在70%~120%之间,重复测定RSD≤20%)。
采用逐级稀释烟叶空白样品中目标分析物的方法测定方法的检出限和定量限,结果(见表3)表明检出限(S/N=3)和定量限(S/N=10)均远低于CORESTA和中国烟草总公司规定的残留水平(80 mg/kg)。

表 3 马来酰肼及其糖苷的回收率、检出限和定量限
LOD:S/N=3; LOQ:S/N=10.
2.6 应用
将所建立的方法应用于烟叶中马来酰肼的残留检测和马来酰肼在烟叶中的代谢过程研究,结果发现,喷洒后第一周即可检测到马来酰肼-O-β-D-葡萄糖苷和马来酰肼-N-β-D-葡萄糖苷的生成;随着喷洒后时间的延长,糖苷含量持续上升,马来酰肼残留量则持续降低(见图6)。但是,马来酰肼糖苷的增加量远小于马来酰肼的减少量。以喷洒后第28天为例,此时马来酰肼的含量已由喷洒时的305.0 mg/kg降至56.8 mg/kg,即有246.4 mg/kg(占喷洒总量的80.8%)的马来酰肼被代谢或发生了转移。而马来酰肼糖苷的含量此时为46.1 mg/kg,折合成马来酰肼的含量仅为18.8 mg/kg,即减少的马来酰肼仅有7.6%转化成了马来酰肼糖苷。由于喷洒实验时马来酰肼只被喷洒至烟叶表面,大量损失的马来酰肼可能转移至根、茎等器官或经根部转移至土壤,也可能代谢成了其他物质。
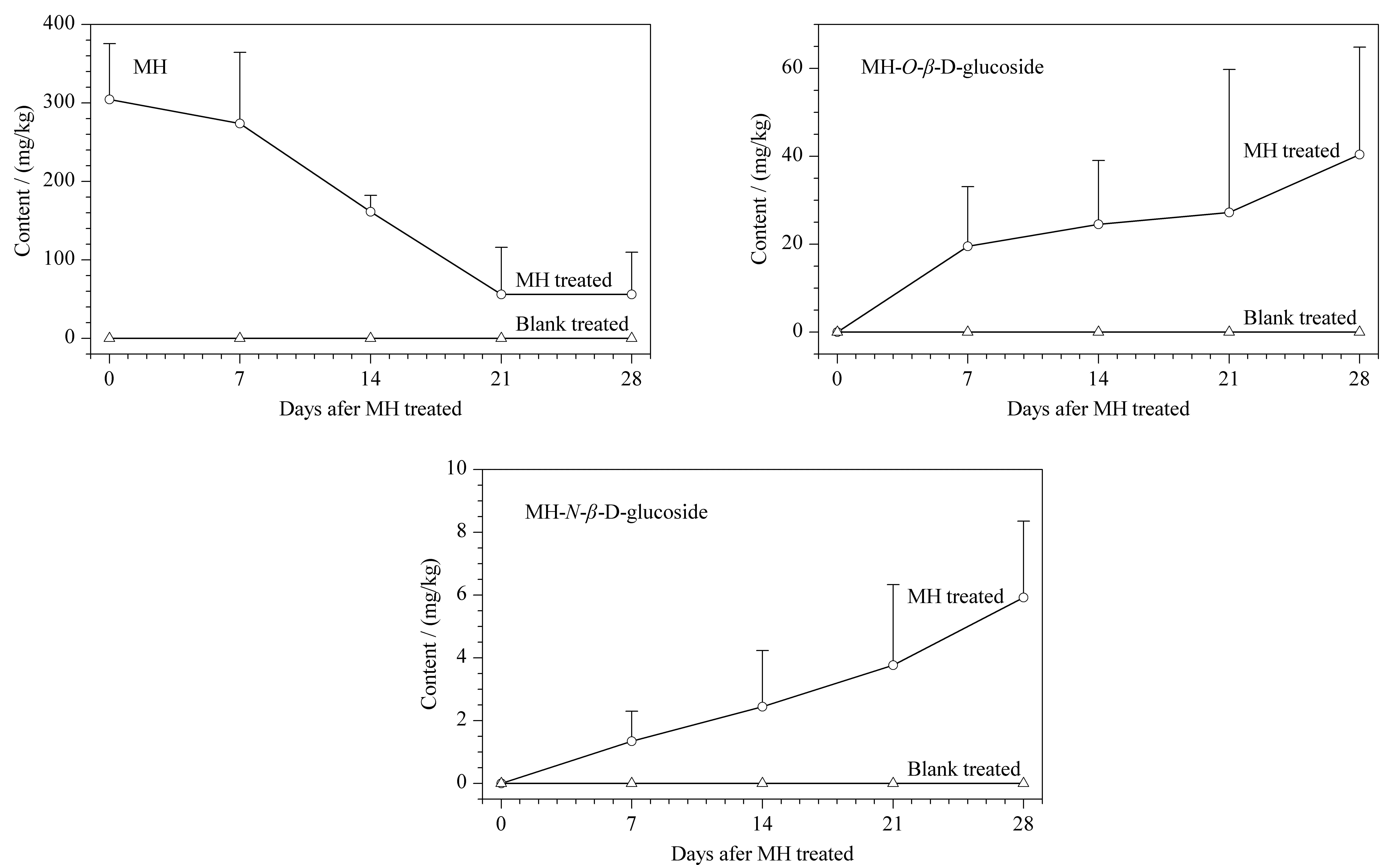
图 6 喷施马来酰肼后烟叶中马来酰肼及其糖苷的含量变化Fig. 6 Content changes of MH and its glucosides in tobacco leaves sprayed with MH
3 结论
本实验建立的马来酰肼及其糖苷的定量分析方法简单、快速、安全、灵敏度高,在样品前处理和仪器分析方面均优于文献方法和烟草行业标准方法。方法的建立对加强烟叶中马来酰肼残留监控和开展马来酰肼代谢相关研究具有重要意义。该方法也可作为其他作物中马来酰肼残留量测定的参考方法。
