多种取代α-卤代酰胺的合成
2018-07-07靳一鸣王夏霖
文 斐,靳一鸣,王夏霖
(湖北科技学院 核技术与化学生物学院,湖北 咸宁 437000)
氮-氧烯丙基正离子是众多1,3-偶极子中一种,二十世纪六十年代,Sheehan在研究α-内酰胺时首次发现并提出了氮-氧烯丙基正离子偶极体[1-3],从此,氮-氧烯丙基正离子偶极体的研究才逐渐被研究人员所认识并得到很好的应用。在后续的研究中发现α-卤代酰胺、α-内酰胺、亚硝基亚烷基卡宾化合物和亚烷基氮氧杂环化合物等都可以经过氮-氧烯丙基正离子偶极体中间体[4]。近年来α-卤代酰胺作为1,3-偶极子参与环合构建杂环化合物的反应已成为有机合成的研究热点之一,例如:Jeffrey, C. S.课题组对α-卤代酰胺的应用中做了巨大的贡献,该课题组先后利用α-卤代酰胺与呋喃类化合物发生氮杂[4+3]偶极环加成反应[5]合成出七元N,O杂环化合物,利用α-卤代酰胺与环戊二烯类化合物发生氮杂[4+3]偶极环加成反应,合成出七元N杂环化合物[6]。Jeffrey, C. S.课题组和Lin, A. J.课题组利用α-卤代酰胺在碱性条件下产生氮-氧烯丙基正离子偶极子与C=O化合物发生[3+2]环加成反应,合成出噁唑啉类化合物[7-8]。Jeffrey, C. S.课题组和Wu, J.课题组同时发现α-卤代酰胺形成的氮-氧烯丙基正离子与吲哚类衍生物发生[3+2]环加成生成吡咯并吲哚类产物[9-10],Liao, X. B.课题组利用这一反应成功合成出天然产物(±)-Minfiensine[11]。Chen, Y. C.课题组研究发现α-卤代酰胺可以与硫叶立德发生[3+1]和[3+2]环加成反应[12];Wu, J.课题组研究发现α-卤代酰胺与异喹啉氮氧化物发生[3+3]环加成反应[13];如果分子既含有α-卤代酰胺又含有呋喃基团时这样的分子在一定条件同样可以发生分子内的[4+3]环加成反应[14-15]。
基于对氮-氧烯丙基正离子反应的研究兴趣,我们课题组已成功将其应用到一些含氮杂环化合物的合成中。如:通过α-卤代酰胺形成氮-氧烯丙基正离子与炔类化合物、硝酮、氯化肟、异氰酸酯和硫代异氰酸酯等偶极环加成反应构建含氮五元、六元杂环化合物[16-17]。
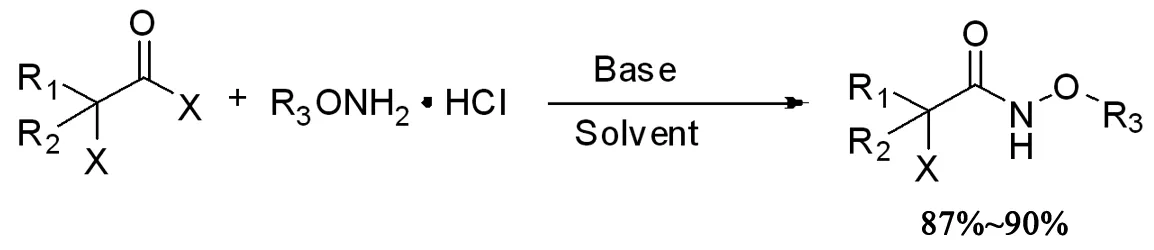
本文将通过下列合成方法合成多种α-卤代酰胺,该方法操作简便,原料易得,产率高达87%~90%。
1 结果与讨论
1.1 反应条件筛选
为了达到最佳的产率,本论文对反应的碱、溶剂、反应温度和反应时间这些反应条件进行了筛选。在反应进行到2h时,产率仅59%,反应未完全。常温下反应产率非常低。在二氯甲烷、乙腈、甲苯和四氢呋喃几种溶剂中,二氯甲烷中产物的产率最高,产率达90%。当把Et3N换成Na2CO3时反应产率下降到64%(表 1中Entry 1,7)。综上所述,在一锅法合成α-卤代酰胺时,采用三乙胺作碱,二氯甲烷作溶剂,在0℃冰水浴中,反应进行到4h时产率最高,达90%。

表1 反应条件筛选
1.2 反应底物拓展
通过改变反应原料连接官能团,拓展反应底物,研究反应的普适性,研究发现反应的普适性比较好,无论是单取的(表2 中Entry 1a)α-卤代酰胺,还是空间位阻较大的(表2 中Entry 1i)α-卤代酰胺都能高产率转化生成。

表2 反应底物拓展

EntryR1R2XR3Yield/%1aMeHBrBn901bMeHBrMe881cMeMeBrBn891dEtHBrBn901eMeHClBn871fHClClBn891gClClClBn881hPhHBrBn891iPhPhClBn871jMeEtBrBn861kCyclopentylHBrBn871lbromomethylHBrBn88
2 实验部分
2.1 仪器与试剂
低温恒温反应浴(巩义市华仪仪器),三用紫外分析仪(上海佳鹏科技有限公司ZF-6型),循环水式真空泵(SHZ-D(Ⅲ)),恒温磁力搅拌器(85-2型),红外干燥箱(巩义市英峪予华仪器),调温电热套(KDM型),旋转蒸发仪(RE-52A),分析天平,核磁共振波谱仪(Bruker DPX 400)。
所用试剂均为化学纯或分析纯,所用无水溶剂都按照标准的无水化方法处理所得。
2.2 N-(苄氧基)-2-溴丙酰胺的合成(1a)
在50 mL圆底烧瓶中加入1 g苄氧胺基盐酸盐,溶于20 mL无水二氯甲烷,在0℃冰水浴中逐滴加入0.8 mL的三乙胺,在1min内将2-溴丙酰溴加入,冰水浴条件下反应,TLC检测反应进程,直至原料点完全消失反应结束,淬灭反应。用二氯甲烷萃取三次,无水硫酸钠干燥有机相,减压浓缩即得目标产物N-(苄氧基)-2-溴丙酰胺(1a),白色固体,产率90%;1H NMR(400 MHz, CDCl3) δ:9.25(brs,1H),7.43~7.28(m,5H),4.94(s,2H),4.33~4.31(d,2H),1.85~1.82(m,3H)。

使用类似方法合成以下产物1b~1h,1l。
2-溴-N-甲氧基丙酰胺(1b):淡黄色油状液体,产率88%;1H NMR(400 MHz, CDCl3)δ:11.0(s,1H),4.46(q,1H,J=7.1Hz),3.77(s,3H),1.81(d,3H,J= 7.1Hz);13C NMR(100 MHz, CDCl3)δ:168.0,64.1,40.1,21.7。
N-(苄氧基)-2-溴-2-甲基丙酰胺(1c):白色固体,产率90%;1H NMR(400 MHz,CDCl3)δ:9.09(brs,1H),7.45~7.28(m,5H),4.96(s,2H),1.96(s,6H)。
N-(苄氧基)-2-溴丁酰胺(1d):白色固体,产率90%;1H NMR(400 MHz, CDCl3)δ:9.08(s,1H),7.42~7.40(m,5H),4.95(s,2H),4.14(t,1H,J= 6.0Hz),2.18~2.00(m,2H),1.02(t,3H,J=7.1Hz);13C NMR(100MHz,CDCl3) δ:166.4,134.7,129.5,129.0,128.7,78.3,48.9,28.8,11.9。
N-(苄氧基)-2-氯丙酰胺(1e):白色固体,产率89%;1H NMR (400MHz, CDCl3)δ:8.92(s,1H),7.44~7.40(m,5H),4.97(s,2H),4.38(q,1H,J= 7.1Hz),1.75(d,3H,J= 7.1Hz);13C NMR(100MHz,CDCl3)δ:167.0,134.7,129.4,129.0,128.7,78.4,53.0,22.2。
N-(苄氧基)-2,2-二氯乙酰胺(1f):白色固体,产率90%;1H NMR(400 MHz,CDCl3)δ:7.43~7.28(t,5H),5.97(s,1H),4.99(s,2H)。
N-(苄氧基)-2,2,2-三氯乙酰胺(1g):淡黄色固体,产率90%;1H NMR(400 MHz,CDCl3)δ:9.23(brs,1H),7.47~7.28(m,6H),5.03(s,2H)。
N-(苄氧基)-2-溴-2-苯基乙酰胺(1h):白色固体,产率89%;1H NMR (400MHz, CDCl3)δ:9.80(s,1H),7.50~7.33(m,10 h),5.30(s,1H),4.92(s,2H)。
N-(苄氧基)-1,2-二溴丙酰胺(1l):淡黄色油状液体,产率88%,1H NMR(400 MHz,CDCl3)δ:7.43(s,5H),4.99(s,2H),4.25(s,1H),4.06~4.01(t,1H),3.77~3.71(t,1H)。
2.3 N-(苄氧基)-2-氯-2,2-二苯基乙酰胺的合成(1i)

在50 mL圆底烧瓶中加入2-羟基-2,2-二苯基乙酸(1 g)与乙酰氯(0.42 g)溶于20 mL的无水二氯甲烷中,室温条件下反应,TLC跟踪反应,反应完全后,淬灭反应,用二氯甲烷(15 mL×3)萃取,无水Na2SO4干燥,减压蒸馏除去溶剂得到油状物 2a;再把2a溶解于二氯亚砜(10 mL)中,80℃条件下回流6 h,减压蒸馏除去过量的二氯亚砜得到产物2b;再在0℃冰水浴中把2b逐滴加入到苄氧胺基盐酸盐与三乙胺的二氯甲烷溶液中,冰水浴条件下反应,TLC检测反应进程,直至原料点完全消失反应结束,淬灭反应。用二氯甲烷萃取三次,无水硫酸钠干燥有机相,减压浓缩即得目标产物N-(苄氧基)-2-氯-2,2-二苯基乙酰胺(1i):白色固体,产率87%;1H NMR (400MHz, CDCl3)δ:7.39~7.37(m,15H),5.03(s,1H),4.89(s,2H)。
2.4 N-(苄氧基)-2-溴-2-甲基丁酰胺的合成(1j)
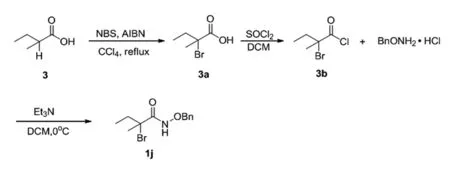
在50 mL的圆底烧瓶中加入2-甲基丁酸(1 g),N-溴代琥珀酰亚胺(2.62 g)和偶氮二异丁腈(0.16 g),溶于无水四氯化碳,80℃冷凝回流反应,TLC跟踪反应,反应完全后,淬灭反应,用二氯甲烷(15 mL×3)萃取,无水Na2SO4干燥,减压蒸馏除去溶剂得到油状物3a,再把3a溶解于二氯亚砜(10 mL)中,80℃条件下回流6h,减压蒸馏除去过量的二氯亚砜得到产物3b;再在0℃冰水浴中把3b逐滴加入到苄氧胺基盐酸盐与三乙胺的二氯甲烷溶液中,冰水浴条件下反应,TLC检测反应进程,直至原料点完全消失反应结束,淬灭反应。用二氯甲烷萃取三次,无水硫酸钠干燥有机相,减压浓缩即得目标产物1j,N-(苄氧基)-2-溴-2-甲基丁酰胺(1j):白色固体,产率86%;1H NMR(400 MHz,CDCl3)δ:9.20(brs,1H),7.46~7.39(m,5H),4.99~4.92(q,2H),2.24~2.18(q,1H),2.20~1.92(q,4H),1.03~1.00(t,3H);13C NMR(400 MHz,CDCl3)δ:168.13,134.75,129.44,128.99,128.67,78.36,77.39,77.07,76.76,67.47,37.13,30.70,10.54。
使用相同的方法合成1k,N-(苄氧基)-1-溴环戊烷甲酰胺(1k):黄棕色固体,产率87%;1H NMR(400 MHz,CDCl3)δ:9.11(brs,1H),7.46~7.28(m,6H),4.97(s,2H),2.47~2.41(m,2H),2.26~2.20(m,2H),2.21~1.97(m,2H),1.92~1.87(m,2H)。
2.5 结论
本文提供了三种α-溴代酰胺的合成方法,经1H-NMR和13C-NMR进行结构测定,产率可达到90%。提供了α-溴代酰胺的合成新思路,该方法操作简便,原料廉价易得,产物的利用度高,可广泛应用于杂环化合物的合成。
[1]Lengyel I,Sheehan J C.α-Lactams (Aziridinones) [J]. Angewandte Chemie International Edtion in English,1968,7:25.
[2]L'Abbe G.Heterocyclic analogues of methylenecyclopropanes[J].Angewandte Chemie International Edtion in English,1980,19:276.
[3]Barnes K L,Koster A K,Jeffrey C S.Trapping the elusive aza-oxyallylic cation: new opportunities in heterocycloaddition chemistry[J].Tetrahedron Letters,2014,55:4690.
[4] Cohen A D,Showalter B M,Toscano J P. Time-resolved IR detection and study of an iminooxirane intermediate[J]. Organic Letters,2004, 6:401.
[5] Fishman J M,Kiessling L L.Synthesis of functionalizable and degradable polymers by ring opening metathesis polymerization[J].Angewandte Chemie-International Edition,2013, 52:5061.
[6]Jeffrey C S,Barnes K L,Eickhoff J A,et al.Generation and reactivity of aza-oxyallyl cationic intermediates: Aza-[4+3] cycloaddition reactions for heterocycle synthesis[J]. Journal of the American Chemical Society,2011,133:7688.
[7] Zhang K F, Yang C, Yao H Q,et al. [3+2] Cycloaddition reaction of in situ formed azaoxyallyl cations with aldehydes: an approach to oxazolidin-4-ones[J].Organic Letters, 2016, 18: 4618.
[8]Acharya A,Montes K,Jeffrey C S.Access to 4-oxazolidinones:A(3+2) cycloaddition approach[J]. Organic Letters, 2016, 18:6082.
[9] Acharya A, Anumandla D and Jeffrey C S. Dearomative indole cycloaddition reactions of aza-oxyallyl cationic intermediates: modular access to pyrroloindolines[J].Journal of the American Chemical Society,2015, 137:14858.
[10]DiPoto M C,Hughes R P,Wu J. Dearomative indole (3+2) reactions with azaoxyallyl cations - new method for the synthesis of pyrroloindolines[J].Journal of the American Chemical Society, 2015, 137:14861.
[11] Ji W Z,Yao L C,Liao X B. Dearomative indole (3+2) reactions with azaoxyallyl cations - new method for the synthesis of pyrroloindolines[J]. Organic Letters, 2016, 18:628.
[12] Li C,Jiang K,Ouyang Q,et al. [3+1]- and [3+2]-cycloadditions of azaoxyallyl cations and sulfur ylides [J]. Organic Letters, 2016, 18:2738.
[13] An Y Y,Xia H G,Wu J.Base-controlled [3+3] cycloaddition of isoquinoline N-oxides with azaoxyallyl cations[J].Chemical Communications,2016,52:10415.
[14]Acharya A,Eickhoff J A,Jeffrey C S. Intramolecular aza-[4+3] cycloaddition reactions of α-halohydroxamates[J]. Synthesis,2013,45:1825.
[15]Acharya A,Eickhoff J A,Chen K,et al.Access to bicyclic hydroxamate macrocycles via intramolecular aza-(4+3) cyloaddition reactions of aza-oxyallylic cation intermediates[J]. Organic Chemistry Frontiers,2016,3:330.
[16]Wang G Q,Chen R X,Wu M H,et al.Efficient one-pot synthesis of 1,3-dihydro-2H-pyrrol-2-one derivatives via aza-oxyallylic cations[J].Tetrahedron Letters,2017,58: 847.
[17]Wang G Q,Zhao S,Chen R X,et al.Synthesis of thiazolidin-4-ones via [3+2] cycloaddition of in situ generated aza-oxyallylic cations with isothiocyanates[J]. Tetrahedron Letters,2017, 58:4308.
